{"title":"Sweet google O’ mine—The importance of online search engines for MS-facilitated, database-independent identification of peptide-encoded book prefaces","authors":"Alexander Hogrebe, Rosa R. Jersie-Christensen","doi":"10.1016/j.euprot.2019.07.012","DOIUrl":null,"url":null,"abstract":"<div><p>In the recent year, we felt like we were not truly showing our full potential in our PhD projects, and so we were very happy and excited when YPIC announced the ultimate proteomics challenge. This gave us the opportunity of showing off and procrastinating at the same time:) The challenge was to identify the amino acid sequence of 19 synthetic peptides made up from an English text and then find the book that it came from. For this task we chose to run on an Orbitrap Fusion™ Lumos™ Tribrid™ Mass Spectrometer with two different sensitive MS2 resolutions, each with both HCD and CID fragmentation consecutively. This strategy was chosen because we speculated that multiple MS2 scans at high quality would be beneficial over lower resolution, speed and quantity in the relatively sparse sample. The resulting chromatogram did not reveal 19 sharp distinct peaks and it was not clear to us where to start a manual spectra interpretation. We instead used the de novo option in the MaxQuant software and the resulting output gave us two phrases with words that were specific enough to be searched in the magic Google search engine. Google gave us the name of a very famous physicist, namely Sir Joseph John Thomson, and a reference to his book “Rays of positive electricity” from 1913. We then converted the paragraph we believed to be the right one into a FASTA format and used it with MaxQuant to do a database search. This resulted in 16 perfectly FASTA search-identified peptide sequences, one with a missing PTM and one found as a truncated version. The remaining one was identified within the MaxQuant de novo sequencing results. We thus show in this study that our workflow combining de novo spectra analysis algorithms with an online search engine is ideally suited for all applications where users want to decipher peptide-encoded prefaces of 20th century science books.</p></div>","PeriodicalId":38260,"journal":{"name":"EuPA Open Proteomics","volume":"22 ","pages":"Pages 14-18"},"PeriodicalIF":0.0000,"publicationDate":"2019-03-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1016/j.euprot.2019.07.012","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"EuPA Open Proteomics","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S221296851930008X","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 0
Abstract
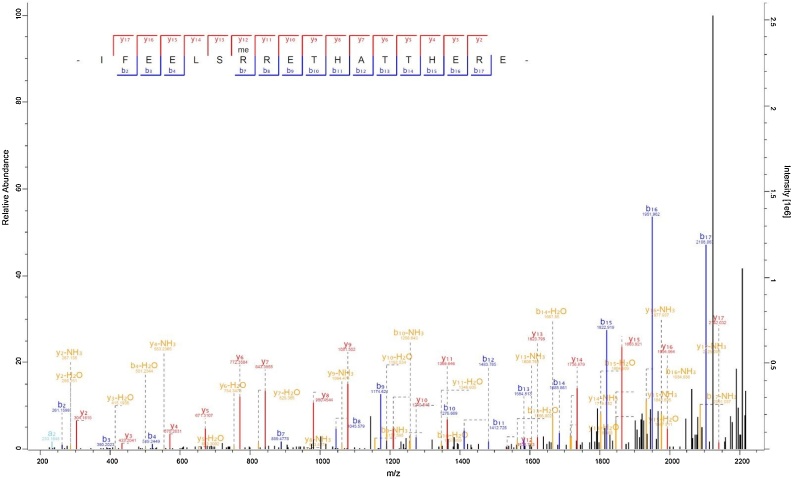
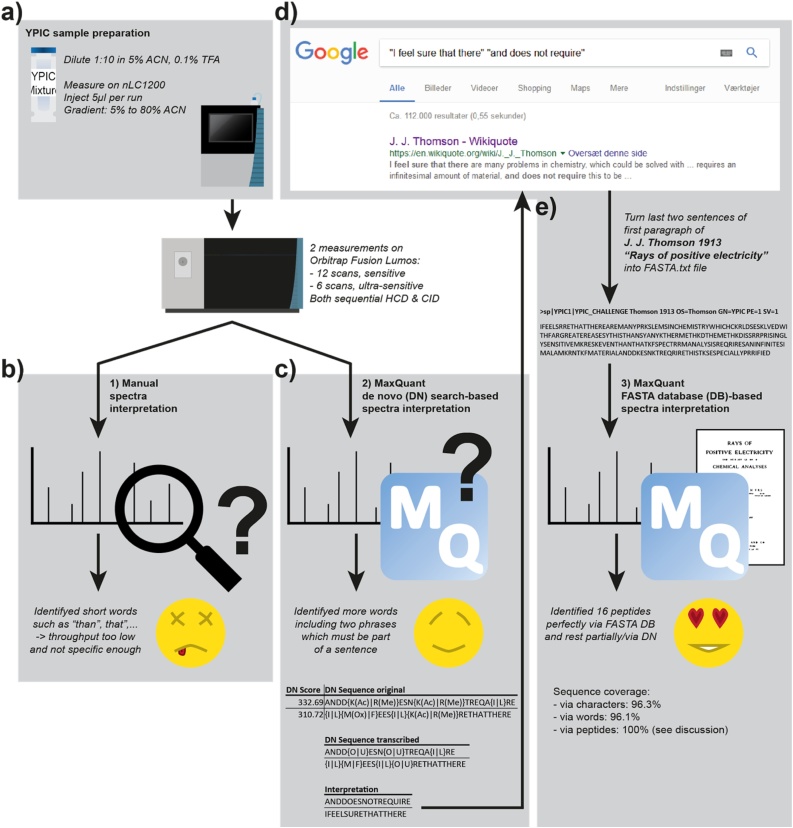
In the recent year, we felt like we were not truly showing our full potential in our PhD projects, and so we were very happy and excited when YPIC announced the ultimate proteomics challenge. This gave us the opportunity of showing off and procrastinating at the same time:) The challenge was to identify the amino acid sequence of 19 synthetic peptides made up from an English text and then find the book that it came from. For this task we chose to run on an Orbitrap Fusion™ Lumos™ Tribrid™ Mass Spectrometer with two different sensitive MS2 resolutions, each with both HCD and CID fragmentation consecutively. This strategy was chosen because we speculated that multiple MS2 scans at high quality would be beneficial over lower resolution, speed and quantity in the relatively sparse sample. The resulting chromatogram did not reveal 19 sharp distinct peaks and it was not clear to us where to start a manual spectra interpretation. We instead used the de novo option in the MaxQuant software and the resulting output gave us two phrases with words that were specific enough to be searched in the magic Google search engine. Google gave us the name of a very famous physicist, namely Sir Joseph John Thomson, and a reference to his book “Rays of positive electricity” from 1913. We then converted the paragraph we believed to be the right one into a FASTA format and used it with MaxQuant to do a database search. This resulted in 16 perfectly FASTA search-identified peptide sequences, one with a missing PTM and one found as a truncated version. The remaining one was identified within the MaxQuant de novo sequencing results. We thus show in this study that our workflow combining de novo spectra analysis algorithms with an online search engine is ideally suited for all applications where users want to decipher peptide-encoded prefaces of 20th century science books.
最近几年,我们觉得我们在博士项目中并没有真正发挥出我们的全部潜力,所以当YPIC宣布最终的蛋白质组学挑战时,我们非常高兴和兴奋。这给了我们一个展示和拖延的机会:)挑战是确定由英文文本组成的19个合成肽的氨基酸序列,然后找到它来自的书。对于这项任务,我们选择在Orbitrap Fusion™Lumos™Tribrid™质谱仪上运行,该质谱仪具有两种不同的灵敏度MS2分辨率,每种都连续具有HCD和CID碎片。之所以选择这种策略,是因为我们推测在相对稀疏的样本中,高质量的多次MS2扫描比低分辨率、低速度和低数量的扫描更有益。得到的色谱图没有显示出19个清晰的峰,我们不清楚从哪里开始手工光谱解释。相反,我们使用了MaxQuant软件中的de novo选项,结果输出给我们两个短语,其中的单词足够具体,可以在神奇的谷歌搜索引擎中搜索。谷歌给了我们一个非常著名的物理学家的名字,即约瑟夫·约翰·汤姆森爵士,以及他1913年出版的《正电射线》一书的参考资料。然后,我们将我们认为正确的段落转换为FASTA格式,并将其与MaxQuant一起进行数据库搜索。这产生了16个完全通过FASTA搜索识别的肽序列,其中一个缺失PTM,另一个被截断。剩余的一个是在MaxQuant de novo测序结果中确定的。因此,我们在这项研究中表明,我们的工作流程将从头开始的光谱分析算法与在线搜索引擎相结合,非常适合用户想要破译20世纪科学书籍的肽编码序言的所有应用。


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


