Cystic Fibrosis Transmembrane Conductance Regulator Genotype, Not Circulating Catecholamines, Influences Cardiovascular Function in Patients with Cystic Fibrosis.
Alexander L Bisch, Courtney M Wheatley, Sarah E Baker, Elizabeth R Peitzman, Erik H Van Iterson, Theresa A Laguna, Wayne J Morgan, Eric M Snyder
{"title":"Cystic Fibrosis Transmembrane Conductance Regulator Genotype, Not Circulating Catecholamines, Influences Cardiovascular Function in Patients with Cystic Fibrosis.","authors":"Alexander L Bisch, Courtney M Wheatley, Sarah E Baker, Elizabeth R Peitzman, Erik H Van Iterson, Theresa A Laguna, Wayne J Morgan, Eric M Snyder","doi":"10.1177/1179548419835788","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Cystic fibrosis (CF) is a genetic disease affecting multiple organ systems of the body and is characterized by mutation in the gene coding for the cystic fibrosis transmembrane conductance regulator (CFTR). Previous work has shown that a single dose of aβ-agonist increases cardiac output (Q) and stroke volume (SV) and decreases systemic vascular resistance (SVR) in healthy subjects. This effect is attenuated in patients with CF; however, the mechanism is unknown. Potential explanations for this decreased cardiovascular response to a β-agonist in CF include inherent cardiovascular deficits secondary to the CFTR mutation, receptor desensitization from prolonged β-agonist use as part of clinical care, or inhibited drug delivery to the bloodstream due to mucus buildup in the lungs. This study sought to determine the effects of endogenous epinephrine (EPI) and norepinephrine (NE) on cardiovascular function in CF and to evaluate the relationship between cardiovascular function and CFTR F508del mutation.</p><p><strong>Methods: </strong>A total of 19 patients with CF and 31 healthy control subjects completed an assessment of Q (C<sub>2</sub>H<sub>2</sub> rebreathing), SV (calculated from Q and heart rate [HR]), Q and SV indexed to body surface area (BSA, QI, and SVI, respectively), SVR (through assessment of Q and mean arterial blood pressure [MAP]), and HR (from 12-lead electrocardiogram [ECG]) at rest along with plasma measures of EPI and NE. We compared subjects by variables of cardiovascular function relative to EPI and NE, and also based on genetic variants of the F508del mutation (homozygous deletion for F508del, heterozygous deletion for F508del, or no deletion of F508del).</p><p><strong>Results: </strong>Cystic fibrosis patients demonstrated significantly lower BSA (CF = 1.71 ± 0.05 m<sup>2</sup> vs healthy = 1.84 ± 0.04 m<sup>2</sup>, <i>P</i> = .03) and SVI (CF = 30.6 ± 2.5 mL/beat/m<sup>2</sup> vs healthy = 39.9 ± 2.5 mL/beat/m<sup>2</sup>, <i>P</i> = .02) when compared with healthy subjects. Cystic fibrosis patients also demonstrated lower Q (CF = 4.58 ± 0.36 L/min vs healthy = 5.71 ± 0.32 L/min, <i>P</i> = .03) and SV (CF = 54 ± 5.5 mL/beat vs healthy = 73.3 ± 4.5 mL/beat, <i>P</i> = .01), and a higher HR (CF = 93.2 ± 3.9 bpm vs healthy = 80.5 ± 2.7 bpm, <i>P</i> < .01) and SVR (CF = 2082 ± 156 dynes*s/cm<sup>-5</sup> vs healthy = 1616 ± 74 dynes*s/cm<sup>-5</sup>, <i>P</i> = .01) compared with healthy subjects. Furthermore, CF patients demonstrated a lower SV (<i>P</i> < .01) corrected for NE when compared with healthy subjects. No significant differences were seen in HR or Q relative to NE, or SVR relative to EPI. Differences were seen in SV (F<sub>(2,14)</sub> = 7.982, <i>P</i> < .01) and SV index (F<sub>(2,14)</sub> = 2.913, <i>P</i> = .08) when patients with CF were stratified according to F508del mutation (number of deletions).</p><p><strong>Conclusions: </strong>Individuals with CF have lower cardiac and peripheral hemodynamic function parameters at rest. Furthermore, these results suggest that impairment in cardiovascular function is likely the result of F508del CFTR genotype, rather than receptor desensitization or inhibited drug delivery.</p>","PeriodicalId":44269,"journal":{"name":"Clinical Medicine Insights-Circulatory Respiratory and Pulmonary Medicine","volume":"13 ","pages":"1179548419835788"},"PeriodicalIF":1.0000,"publicationDate":"2019-03-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1177/1179548419835788","citationCount":"5","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Medicine Insights-Circulatory Respiratory and Pulmonary Medicine","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/1179548419835788","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2019/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"RESPIRATORY SYSTEM","Score":null,"Total":0}
引用次数: 5
Abstract
Background: Cystic fibrosis (CF) is a genetic disease affecting multiple organ systems of the body and is characterized by mutation in the gene coding for the cystic fibrosis transmembrane conductance regulator (CFTR). Previous work has shown that a single dose of aβ-agonist increases cardiac output (Q) and stroke volume (SV) and decreases systemic vascular resistance (SVR) in healthy subjects. This effect is attenuated in patients with CF; however, the mechanism is unknown. Potential explanations for this decreased cardiovascular response to a β-agonist in CF include inherent cardiovascular deficits secondary to the CFTR mutation, receptor desensitization from prolonged β-agonist use as part of clinical care, or inhibited drug delivery to the bloodstream due to mucus buildup in the lungs. This study sought to determine the effects of endogenous epinephrine (EPI) and norepinephrine (NE) on cardiovascular function in CF and to evaluate the relationship between cardiovascular function and CFTR F508del mutation.
Methods: A total of 19 patients with CF and 31 healthy control subjects completed an assessment of Q (C2H2 rebreathing), SV (calculated from Q and heart rate [HR]), Q and SV indexed to body surface area (BSA, QI, and SVI, respectively), SVR (through assessment of Q and mean arterial blood pressure [MAP]), and HR (from 12-lead electrocardiogram [ECG]) at rest along with plasma measures of EPI and NE. We compared subjects by variables of cardiovascular function relative to EPI and NE, and also based on genetic variants of the F508del mutation (homozygous deletion for F508del, heterozygous deletion for F508del, or no deletion of F508del).
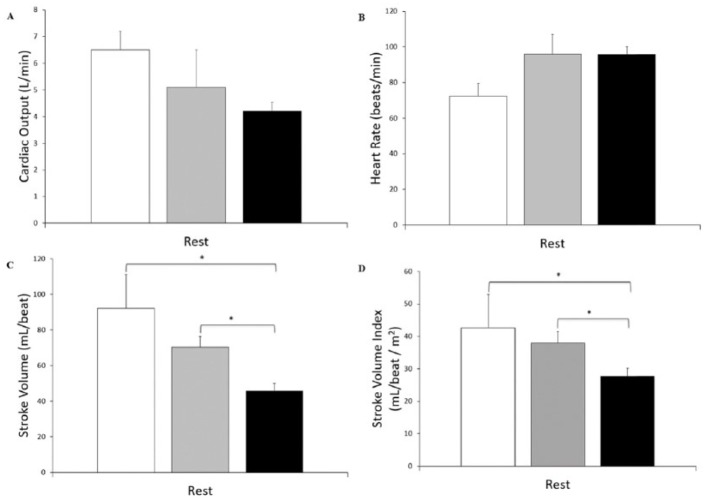
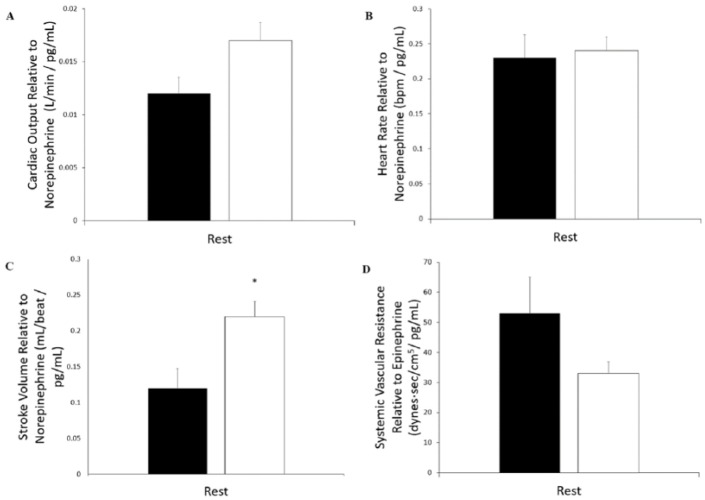
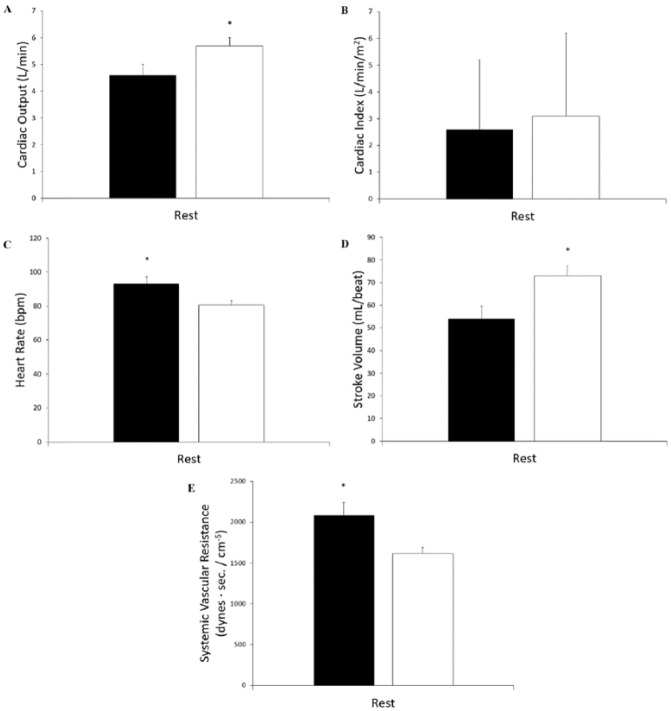
Results: Cystic fibrosis patients demonstrated significantly lower BSA (CF = 1.71 ± 0.05 m2 vs healthy = 1.84 ± 0.04 m2, P = .03) and SVI (CF = 30.6 ± 2.5 mL/beat/m2 vs healthy = 39.9 ± 2.5 mL/beat/m2, P = .02) when compared with healthy subjects. Cystic fibrosis patients also demonstrated lower Q (CF = 4.58 ± 0.36 L/min vs healthy = 5.71 ± 0.32 L/min, P = .03) and SV (CF = 54 ± 5.5 mL/beat vs healthy = 73.3 ± 4.5 mL/beat, P = .01), and a higher HR (CF = 93.2 ± 3.9 bpm vs healthy = 80.5 ± 2.7 bpm, P < .01) and SVR (CF = 2082 ± 156 dynes*s/cm-5 vs healthy = 1616 ± 74 dynes*s/cm-5, P = .01) compared with healthy subjects. Furthermore, CF patients demonstrated a lower SV (P < .01) corrected for NE when compared with healthy subjects. No significant differences were seen in HR or Q relative to NE, or SVR relative to EPI. Differences were seen in SV (F(2,14) = 7.982, P < .01) and SV index (F(2,14) = 2.913, P = .08) when patients with CF were stratified according to F508del mutation (number of deletions).
Conclusions: Individuals with CF have lower cardiac and peripheral hemodynamic function parameters at rest. Furthermore, these results suggest that impairment in cardiovascular function is likely the result of F508del CFTR genotype, rather than receptor desensitization or inhibited drug delivery.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


