Júlia Guasti P Vianna, Thiago Gabriel Simor, Pamella Senna, Michell Roncete De Bortoli, Everlayny Fiorot Costalonga, Antonio Carlos Seguro, Weverton Machado Luchi
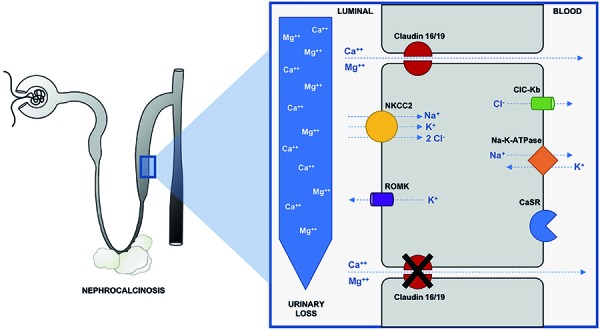
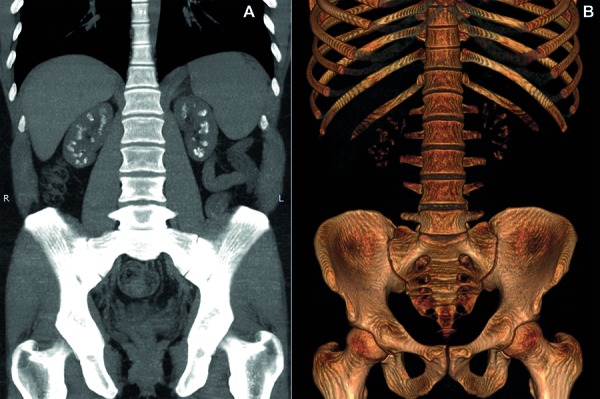
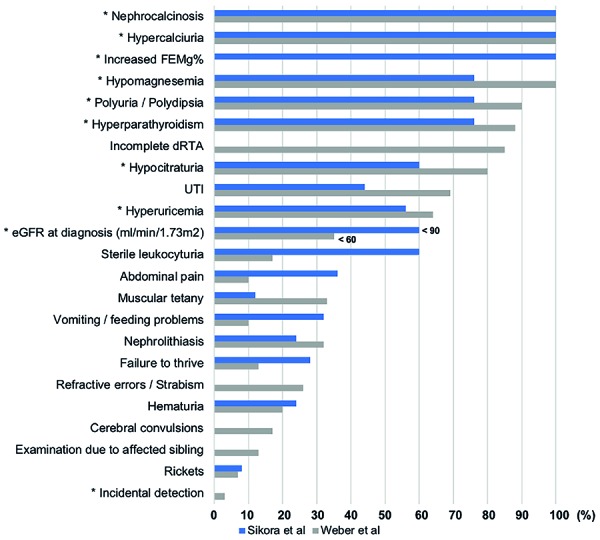
{"title":"Atypical presentation of familial hypomagnesemia with hypercalciuria and nephrocalcinosis in a patient with a new claudin-16 gene mutation.","authors":"Júlia Guasti P Vianna, Thiago Gabriel Simor, Pamella Senna, Michell Roncete De Bortoli, Everlayny Fiorot Costalonga, Antonio Carlos Seguro, Weverton Machado Luchi","doi":"10.5414/CNCS109595","DOIUrl":null,"url":null,"abstract":"<p><p>Familial hypomagnesemia with hypercalciuria and nephrocalcinosis (FHHNC) is an autosomal recessive tubular disorder caused by mutations in genes that encode renal tight junction proteins claudin-16 or claudin-19, which are responsible for magnesium and calcium paracellular reabsorption in the thick ascending limb of Henle's loop. Progressive renal failure is frequently present, and most of the patients require renal replacement therapy still during adolescence. In this case report, we describe a new homozygous missense mutation on <i>CLDN16</i> gene (c.592G>C, Gly198Arg) in a 24-year-old male patient diagnosed with FHHNC after clinical investigation due to incidental detection of altered routine laboratorial tests, who was firstly misdiagnosed with primary hyperparathyroidism. In addition, it illustrates an atypical presentation of this disease, with late onset of chronic kidney disease, improving the phenotype-genotype knowledge of patients with FHHNC.</p>","PeriodicalId":10398,"journal":{"name":"Clinical Nephrology. Case Studies","volume":"7 ","pages":"27-34"},"PeriodicalIF":0.0000,"publicationDate":"2019-05-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.5414/CNCS109595","citationCount":"6","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Nephrology. Case Studies","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.5414/CNCS109595","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2019/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 6
Abstract
Familial hypomagnesemia with hypercalciuria and nephrocalcinosis (FHHNC) is an autosomal recessive tubular disorder caused by mutations in genes that encode renal tight junction proteins claudin-16 or claudin-19, which are responsible for magnesium and calcium paracellular reabsorption in the thick ascending limb of Henle's loop. Progressive renal failure is frequently present, and most of the patients require renal replacement therapy still during adolescence. In this case report, we describe a new homozygous missense mutation on CLDN16 gene (c.592G>C, Gly198Arg) in a 24-year-old male patient diagnosed with FHHNC after clinical investigation due to incidental detection of altered routine laboratorial tests, who was firstly misdiagnosed with primary hyperparathyroidism. In addition, it illustrates an atypical presentation of this disease, with late onset of chronic kidney disease, improving the phenotype-genotype knowledge of patients with FHHNC.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


