Yangyang Hao, Quan-Yang Duh, Richard T Kloos, Joshua Babiarz, R Mack Harrell, S Thomas Traweek, Su Yeon Kim, Grazyna Fedorowicz, P Sean Walsh, Peter M Sadow, Jing Huang, Giulia C Kennedy
{"title":"Identification of Hürthle cell cancers: solving a clinical challenge with genomic sequencing and a trio of machine learning algorithms.","authors":"Yangyang Hao, Quan-Yang Duh, Richard T Kloos, Joshua Babiarz, R Mack Harrell, S Thomas Traweek, Su Yeon Kim, Grazyna Fedorowicz, P Sean Walsh, Peter M Sadow, Jing Huang, Giulia C Kennedy","doi":"10.1186/s12918-019-0693-z","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Identification of Hürthle cell cancers by non-operative fine-needle aspiration biopsy (FNAB) of thyroid nodules is challenging. Resultingly, non-cancerous Hürthle lesions were conventionally distinguished from Hürthle cell cancers by histopathological examination of tissue following surgical resection. Reliance on histopathological evaluation requires patients to undergo surgery to obtain a diagnosis despite most being non-cancerous. It is highly desirable to avoid surgery and to provide accurate classification of benignity versus malignancy from FNAB preoperatively. In our first-generation algorithm, Gene Expression Classifier (GEC), we achieved this goal by using machine learning (ML) on gene expression features. The classifier is sensitive, but not specific due in part to the presence of non-neoplastic benign Hürthle cells in many FNAB.</p><p><strong>Results: </strong>We sought to overcome this low-specificity limitation by expanding the feature set for ML using next-generation whole transcriptome RNA sequencing and called the improved algorithm the Genomic Sequencing Classifier (GSC). The Hürthle identification leverages mitochondrial expression and we developed novel feature extraction mechanisms to measure chromosomal and genomic level loss-of-heterozygosity (LOH) for the algorithm. Additionally, we developed a multi-layered system of cascading classifiers to sequentially triage Hürthle cell-containing FNAB, including: 1. presence of Hürthle cells, 2. presence of neoplastic Hürthle cells, and 3. presence of benign Hürthle cells. The final Hürthle cell Index utilizes 1048 nuclear and mitochondrial genes; and Hürthle cell Neoplasm Index leverages LOH features as well as 2041 genes. Both indices are Support Vector Machine (SVM) based. The third classifier, the GSC Benign/Suspicious classifier, utilizes 1115 core genes and is an ensemble classifier incorporating 12 individual models.</p><p><strong>Conclusions: </strong>The accurate algorithmic depiction of this complex biological system among Hürthle subtypes results in a dramatic improvement of classification performance; specificity among Hürthle cell neoplasms increases from 11.8% with the GEC to 58.8% with the GSC, while maintaining the same sensitivity of 89%.</p>","PeriodicalId":9013,"journal":{"name":"BMC Systems Biology","volume":null,"pages":null},"PeriodicalIF":0.0000,"publicationDate":"2019-04-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s12918-019-0693-z","citationCount":"24","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Systems Biology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s12918-019-0693-z","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Mathematics","Score":null,"Total":0}
引用次数: 24
Abstract
Background: Identification of Hürthle cell cancers by non-operative fine-needle aspiration biopsy (FNAB) of thyroid nodules is challenging. Resultingly, non-cancerous Hürthle lesions were conventionally distinguished from Hürthle cell cancers by histopathological examination of tissue following surgical resection. Reliance on histopathological evaluation requires patients to undergo surgery to obtain a diagnosis despite most being non-cancerous. It is highly desirable to avoid surgery and to provide accurate classification of benignity versus malignancy from FNAB preoperatively. In our first-generation algorithm, Gene Expression Classifier (GEC), we achieved this goal by using machine learning (ML) on gene expression features. The classifier is sensitive, but not specific due in part to the presence of non-neoplastic benign Hürthle cells in many FNAB.
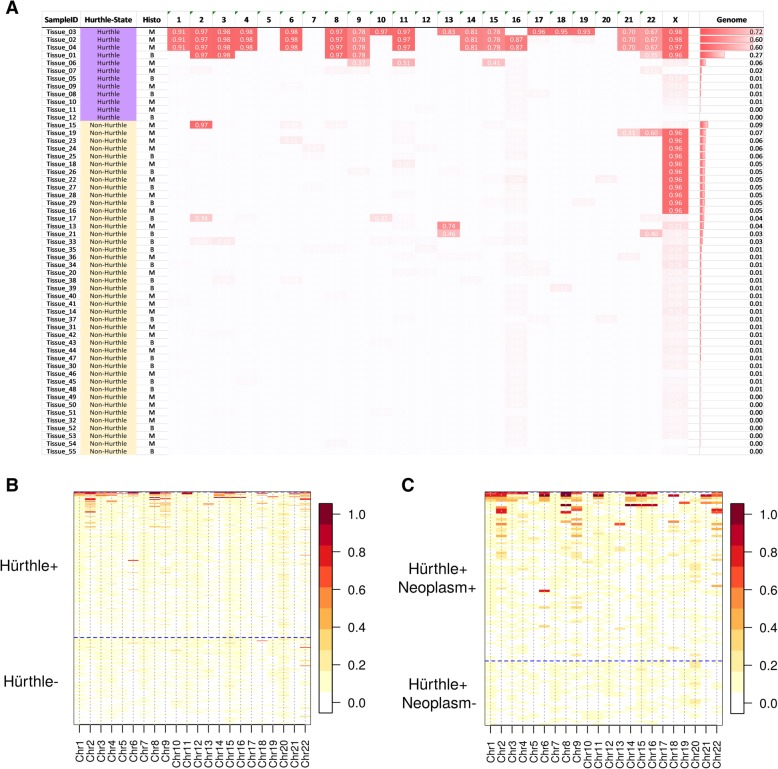
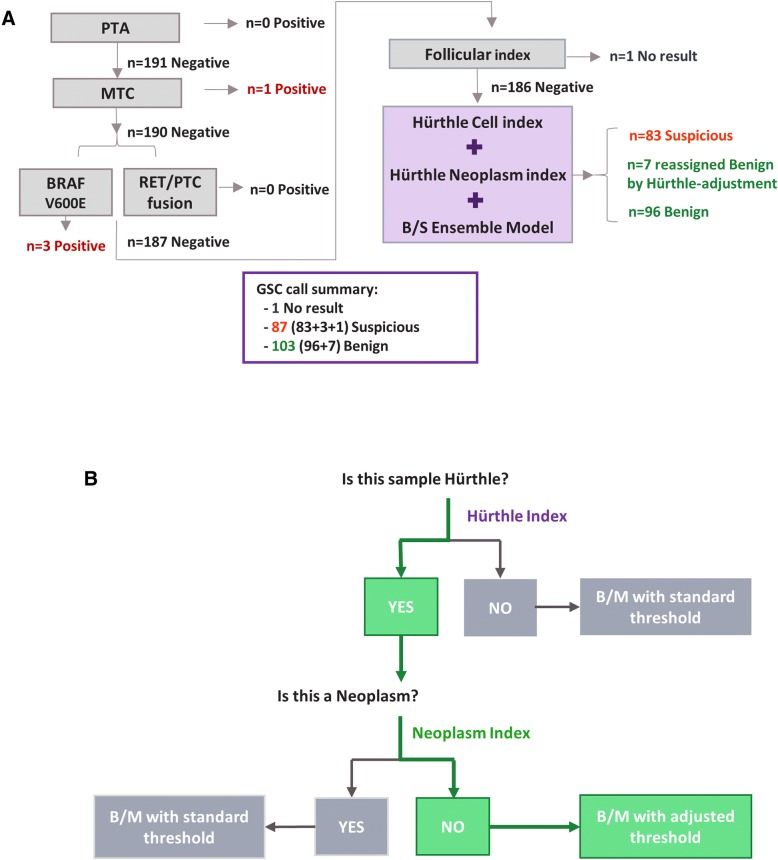
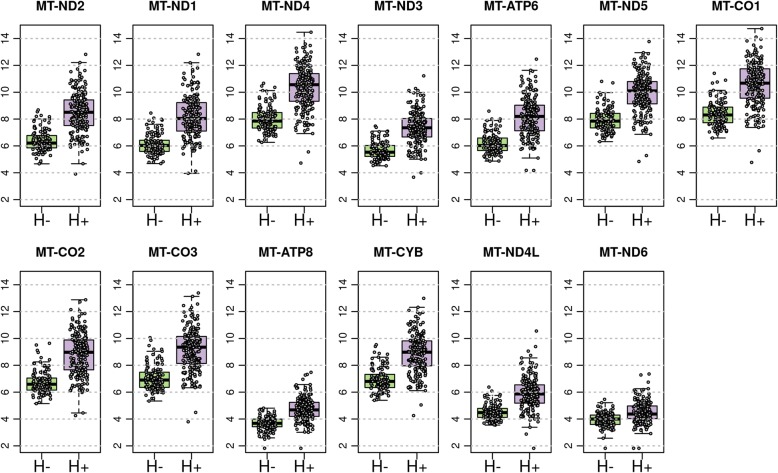
Results: We sought to overcome this low-specificity limitation by expanding the feature set for ML using next-generation whole transcriptome RNA sequencing and called the improved algorithm the Genomic Sequencing Classifier (GSC). The Hürthle identification leverages mitochondrial expression and we developed novel feature extraction mechanisms to measure chromosomal and genomic level loss-of-heterozygosity (LOH) for the algorithm. Additionally, we developed a multi-layered system of cascading classifiers to sequentially triage Hürthle cell-containing FNAB, including: 1. presence of Hürthle cells, 2. presence of neoplastic Hürthle cells, and 3. presence of benign Hürthle cells. The final Hürthle cell Index utilizes 1048 nuclear and mitochondrial genes; and Hürthle cell Neoplasm Index leverages LOH features as well as 2041 genes. Both indices are Support Vector Machine (SVM) based. The third classifier, the GSC Benign/Suspicious classifier, utilizes 1115 core genes and is an ensemble classifier incorporating 12 individual models.
Conclusions: The accurate algorithmic depiction of this complex biological system among Hürthle subtypes results in a dramatic improvement of classification performance; specificity among Hürthle cell neoplasms increases from 11.8% with the GEC to 58.8% with the GSC, while maintaining the same sensitivity of 89%.
期刊介绍:
Cessation.
BMC Systems Biology is an open access journal publishing original peer-reviewed research articles in experimental and theoretical aspects of the function of biological systems at the molecular, cellular or organismal level, in particular those addressing the engineering of biological systems, network modelling, quantitative analyses, integration of different levels of information and synthetic biology.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


