Jiang Zhang, Guangli Wang, Wei-Wu He, Molly Losh, Elizabeth Berry-Kravis, William E Funk
{"title":"Expression and Characterization of Human Fragile X Mental Retardation Protein Isoforms and Interacting Proteins in Human Cells.","authors":"Jiang Zhang, Guangli Wang, Wei-Wu He, Molly Losh, Elizabeth Berry-Kravis, William E Funk","doi":"10.1177/1178641818825268","DOIUrl":null,"url":null,"abstract":"<p><p>Fragile X mental retardation protein is an mRNA-binding protein associated with phenotypic manifestations of fragile X syndrome, an X-linked disorder caused by mutation in the <i>FMR1</i> gene that is the most common inherited cause of intellectual disability. Despite the well-studied genetic mechanism of the disease, the proteoforms of fragile X mental retardation protein have not been thoroughly characterized. Here, we report the expression and mass spectrometric characterization of human fragile X mental retardation protein. <i>FMR1</i> cDNA clone was transfected into human HEK293 cells to express the full-length human fragile X mental retardation protein. Purified fragile X mental retardation protein was subjected to trypsin digestion and characterized by mass spectrometry. Results show 80.5% protein sequence coverage of fragile X mental retardation protein (Q06787, <i>FMR1</i>_HUMAN) including both the N- and C-terminal peptides, indicating successful expression of the full-length protein. Identified post-translational modifications include N-terminal acetylation, phosphorylation (Ser600), and methylation (Arg290, 471, and 474). In addition to the full-length fragile X mental retardation protein isoform (isoform 6), two endogenous fragile X mental retardation protein alternative splicing isoforms (isoforms 4 and 7), as well as fragile X mental retardation protein interacting proteins, were also identified in the co-purified samples, suggesting the interaction network of the human fragile X mental retardation protein. Quantification was performed at the peptide level, and this information provides important reference for the future development of a targeted assay for quantifying fragile X mental retardation protein in clinical samples. Collectively, this study provides the first comprehensive report of human fragile X mental retardation protein proteoforms and may help advance the mechanistic understanding of fragile X syndrome and related phenotypes associated with the <i>FMR1</i> mutation.</p>","PeriodicalId":88975,"journal":{"name":"Proteomics insights","volume":"10 ","pages":"1178641818825268"},"PeriodicalIF":0.0000,"publicationDate":"2019-03-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1177/1178641818825268","citationCount":"4","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Proteomics insights","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/1178641818825268","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2019/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 4
Abstract
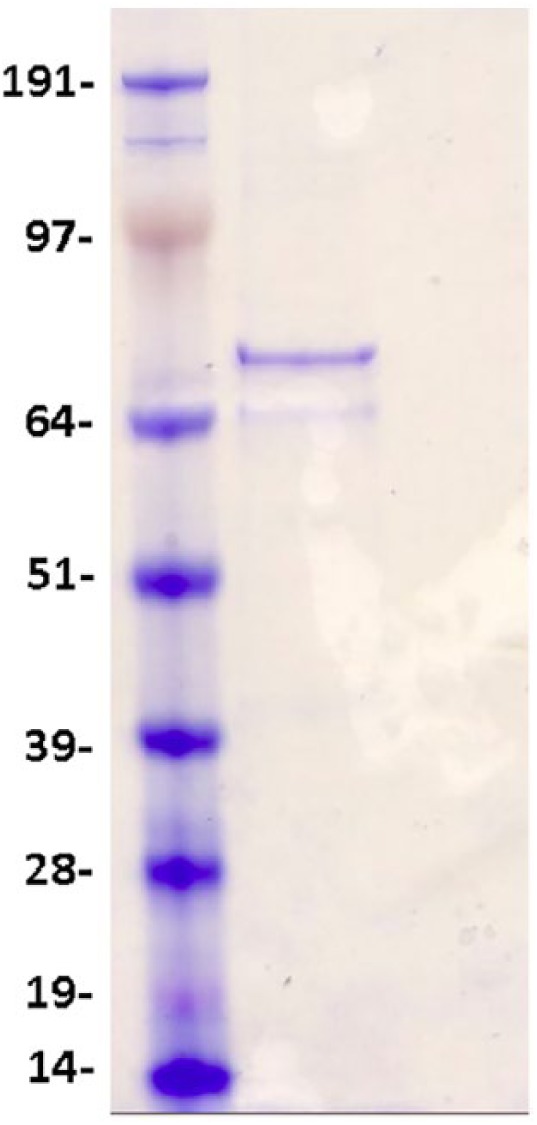
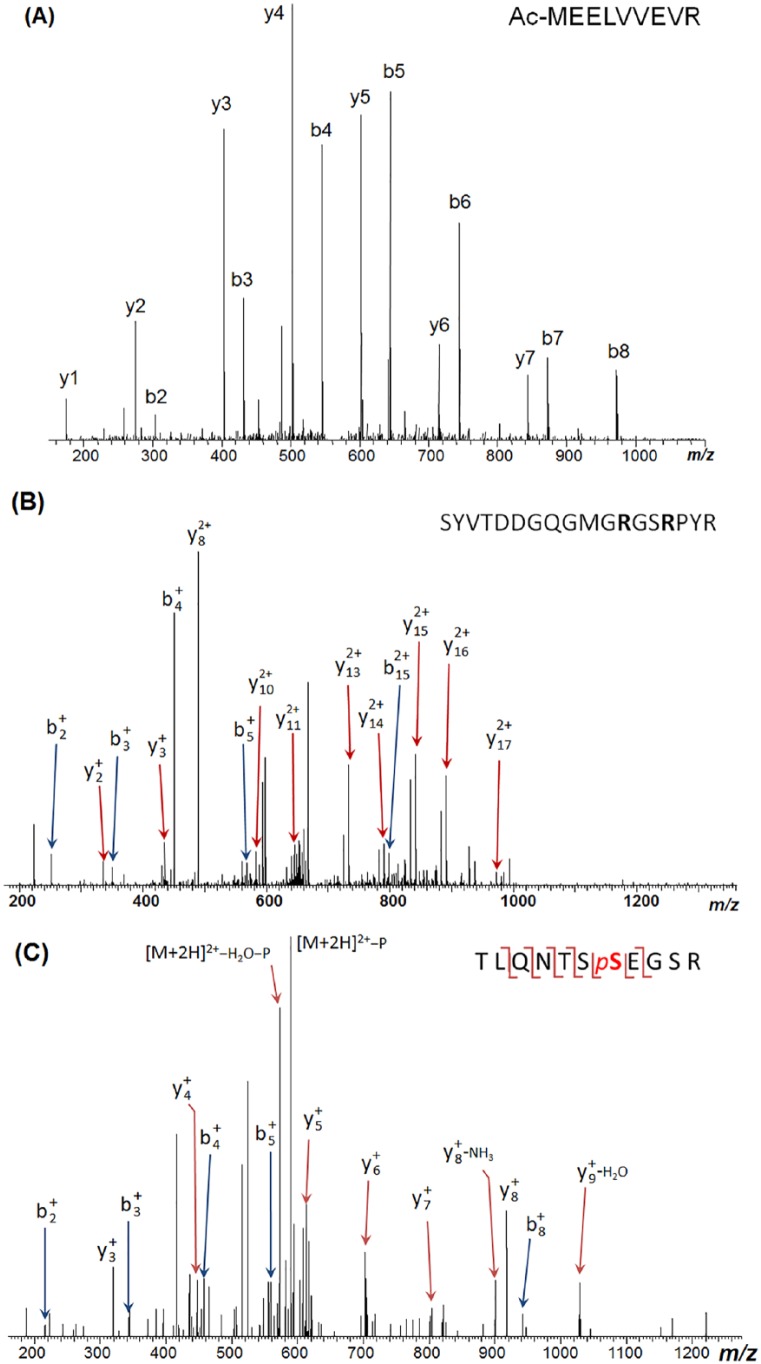
Fragile X mental retardation protein is an mRNA-binding protein associated with phenotypic manifestations of fragile X syndrome, an X-linked disorder caused by mutation in the FMR1 gene that is the most common inherited cause of intellectual disability. Despite the well-studied genetic mechanism of the disease, the proteoforms of fragile X mental retardation protein have not been thoroughly characterized. Here, we report the expression and mass spectrometric characterization of human fragile X mental retardation protein. FMR1 cDNA clone was transfected into human HEK293 cells to express the full-length human fragile X mental retardation protein. Purified fragile X mental retardation protein was subjected to trypsin digestion and characterized by mass spectrometry. Results show 80.5% protein sequence coverage of fragile X mental retardation protein (Q06787, FMR1_HUMAN) including both the N- and C-terminal peptides, indicating successful expression of the full-length protein. Identified post-translational modifications include N-terminal acetylation, phosphorylation (Ser600), and methylation (Arg290, 471, and 474). In addition to the full-length fragile X mental retardation protein isoform (isoform 6), two endogenous fragile X mental retardation protein alternative splicing isoforms (isoforms 4 and 7), as well as fragile X mental retardation protein interacting proteins, were also identified in the co-purified samples, suggesting the interaction network of the human fragile X mental retardation protein. Quantification was performed at the peptide level, and this information provides important reference for the future development of a targeted assay for quantifying fragile X mental retardation protein in clinical samples. Collectively, this study provides the first comprehensive report of human fragile X mental retardation protein proteoforms and may help advance the mechanistic understanding of fragile X syndrome and related phenotypes associated with the FMR1 mutation.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


