{"title":"SNPs detection by eBWT positional clustering.","authors":"Nicola Prezza, Nadia Pisanti, Marinella Sciortino, Giovanna Rosone","doi":"10.1186/s13015-019-0137-8","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Sequencing technologies keep on turning cheaper and faster, thus putting a growing pressure for data structures designed to efficiently store raw data, and possibly perform analysis therein. In this view, there is a growing interest in alignment-free and reference-free variants calling methods that only make use of (suitably indexed) raw reads data.</p><p><strong>Results: </strong>We develop the <i>positional clustering</i> theory that (i) describes how the extended Burrows-Wheeler Transform (eBWT) of a collection of reads tends to cluster together bases that cover the same genome position (ii) predicts the size of such clusters, and (iii) exhibits an elegant and precise LCP array based procedure to locate such clusters in the eBWT. Based on this theory, we designed and implemented an alignment-free and reference-free SNPs calling method, and we devised a consequent SNPs calling pipeline. Experiments on both synthetic and real data show that SNPs can be detected with a simple scan of the eBWT and LCP arrays as, in accordance with our theoretical framework, they are within clusters in the eBWT of the reads. Finally, our tool intrinsically performs a reference-free evaluation of its accuracy by returning the coverage of each SNP.</p><p><strong>Conclusions: </strong>Based on the results of the experiments on synthetic and real data, we conclude that the positional clustering framework can be effectively used for the problem of identifying SNPs, and it appears to be a promising approach for calling other type of variants directly on raw sequencing data.</p><p><strong>Availability: </strong>The software ebwt2snp is freely available for academic use at: https://github.com/nicolaprezza/ebwt2snp.</p>","PeriodicalId":50823,"journal":{"name":"Algorithms for Molecular Biology","volume":" ","pages":"3"},"PeriodicalIF":1.5000,"publicationDate":"2019-02-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s13015-019-0137-8","citationCount":"21","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Algorithms for Molecular Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13015-019-0137-8","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2019/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 21
Abstract
Background: Sequencing technologies keep on turning cheaper and faster, thus putting a growing pressure for data structures designed to efficiently store raw data, and possibly perform analysis therein. In this view, there is a growing interest in alignment-free and reference-free variants calling methods that only make use of (suitably indexed) raw reads data.
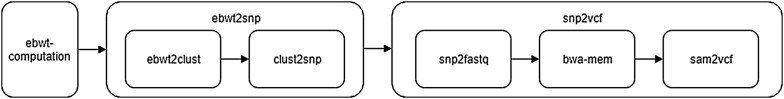
Results: We develop the positional clustering theory that (i) describes how the extended Burrows-Wheeler Transform (eBWT) of a collection of reads tends to cluster together bases that cover the same genome position (ii) predicts the size of such clusters, and (iii) exhibits an elegant and precise LCP array based procedure to locate such clusters in the eBWT. Based on this theory, we designed and implemented an alignment-free and reference-free SNPs calling method, and we devised a consequent SNPs calling pipeline. Experiments on both synthetic and real data show that SNPs can be detected with a simple scan of the eBWT and LCP arrays as, in accordance with our theoretical framework, they are within clusters in the eBWT of the reads. Finally, our tool intrinsically performs a reference-free evaluation of its accuracy by returning the coverage of each SNP.
Conclusions: Based on the results of the experiments on synthetic and real data, we conclude that the positional clustering framework can be effectively used for the problem of identifying SNPs, and it appears to be a promising approach for calling other type of variants directly on raw sequencing data.
Availability: The software ebwt2snp is freely available for academic use at: https://github.com/nicolaprezza/ebwt2snp.
期刊介绍:
Algorithms for Molecular Biology publishes articles on novel algorithms for biological sequence and structure analysis, phylogeny reconstruction, and combinatorial algorithms and machine learning.
Areas of interest include but are not limited to: algorithms for RNA and protein structure analysis, gene prediction and genome analysis, comparative sequence analysis and alignment, phylogeny, gene expression, machine learning, and combinatorial algorithms.
Where appropriate, manuscripts should describe applications to real-world data. However, pure algorithm papers are also welcome if future applications to biological data are to be expected, or if they address complexity or approximation issues of novel computational problems in molecular biology. Articles about novel software tools will be considered for publication if they contain some algorithmically interesting aspects.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


