Chunxiang Fan, Marius Kuhn, Alexander Pepler Mbiol, James Groome, Vern Winston, Saskia Biskup, Frank Lehmann-Horn, Karin Jurkat-Rott
{"title":"Kir2.2 p.Thr140Met: a genetic susceptibility to sporadic periodic paralysis.","authors":"Chunxiang Fan, Marius Kuhn, Alexander Pepler Mbiol, James Groome, Vern Winston, Saskia Biskup, Frank Lehmann-Horn, Karin Jurkat-Rott","doi":"","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>Periodic paralyses (PP) are recurrent episodes of flaccid limb muscle weakness. Next to autosomal dominant forms, sporadic PP (SPP) cases are known but their genetics are unclear.</p><p><strong>Methods: </strong>In a patient with hypokalemic SPP, we performed exome sequencing to identify a candidate gene. We sequenced this gene in 263 unrelated PP patients without any known causative mutations. Then we performed functional analysis of all variants found and molecular modelling for interpretation.</p><p><strong>Results: </strong>Exome sequencing in the proband yielded three heterozygous variants predicted to be linked to disease. These encoded p.Thr140Met in the Kir2.2 potassium channel, p.Asp229Asn in protein kinase C theta, and p.Thr15943Ile in titin. Since all hitherto known causative PP genes code for ion channels, we studied the Kir2.2-encoding gene, <i>KCNJ12</i>, for involvement in PP pathogenesis. <i>KCNJ12</i> screening in 263 PP patients revealed three further variants, each in a single individual and coding for p.Gly419Ser, p.Cys75Tyr, and p.Ile283Val. All four Kir2.2 variants were functionally expressed. Only p.Thr140Met displayed relevant functional alterations, i.e. homo-tetrameric channels produced almost no current, and hetero-tetrameric channels suppressed co-expressed wildtype Kir2.1 in a dominant-negative manner. Molecular modelling showed Kir2.2 p.Thr140Met to reduce movement of potassium ions towards binding sites in the hetero-tetramer pore compatible with a reduced maximal current. MD simulations revealed loss of hydrogen bonding with the p.Thr140Met substitution.</p><p><strong>Discussion: </strong>The electrophysiological findings of p.Thr140Met are similar to those found in thyrotoxic PP caused by Kir2.6 mutations. Also, the homologous Thr140 residue is mutated in Kir2.6. This supports the idea that Kir2.2 p.Thr140Met conveys susceptibility to SPP and should be included in genetic screening.</p>","PeriodicalId":35953,"journal":{"name":"Acta Myologica","volume":"37 3","pages":"193-203"},"PeriodicalIF":0.0000,"publicationDate":"2018-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/e1/78/am-2018-03-193.PMC6390110.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Acta Myologica","FirstCategoryId":"1085","ListUrlMain":"","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"Medicine","Score":null,"Total":0}
引用次数: 0
Abstract
Introduction: Periodic paralyses (PP) are recurrent episodes of flaccid limb muscle weakness. Next to autosomal dominant forms, sporadic PP (SPP) cases are known but their genetics are unclear.
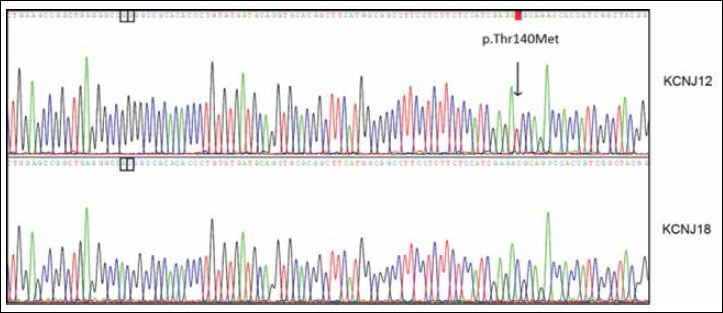
Methods: In a patient with hypokalemic SPP, we performed exome sequencing to identify a candidate gene. We sequenced this gene in 263 unrelated PP patients without any known causative mutations. Then we performed functional analysis of all variants found and molecular modelling for interpretation.
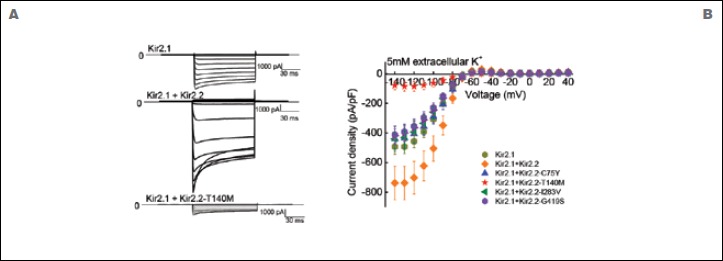
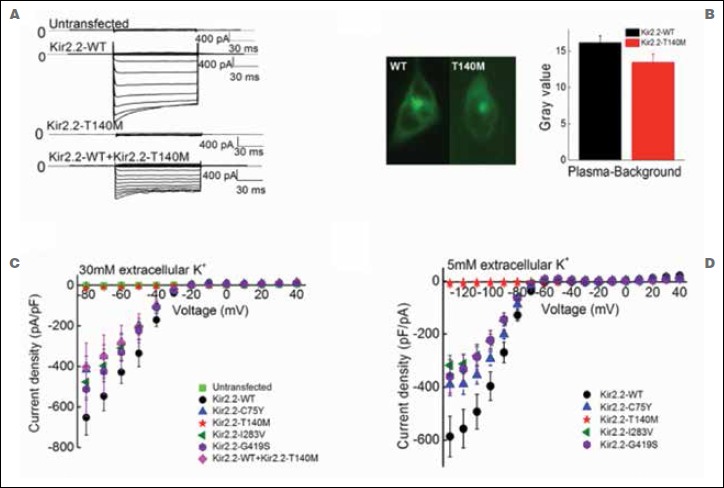
Results: Exome sequencing in the proband yielded three heterozygous variants predicted to be linked to disease. These encoded p.Thr140Met in the Kir2.2 potassium channel, p.Asp229Asn in protein kinase C theta, and p.Thr15943Ile in titin. Since all hitherto known causative PP genes code for ion channels, we studied the Kir2.2-encoding gene, KCNJ12, for involvement in PP pathogenesis. KCNJ12 screening in 263 PP patients revealed three further variants, each in a single individual and coding for p.Gly419Ser, p.Cys75Tyr, and p.Ile283Val. All four Kir2.2 variants were functionally expressed. Only p.Thr140Met displayed relevant functional alterations, i.e. homo-tetrameric channels produced almost no current, and hetero-tetrameric channels suppressed co-expressed wildtype Kir2.1 in a dominant-negative manner. Molecular modelling showed Kir2.2 p.Thr140Met to reduce movement of potassium ions towards binding sites in the hetero-tetramer pore compatible with a reduced maximal current. MD simulations revealed loss of hydrogen bonding with the p.Thr140Met substitution.
Discussion: The electrophysiological findings of p.Thr140Met are similar to those found in thyrotoxic PP caused by Kir2.6 mutations. Also, the homologous Thr140 residue is mutated in Kir2.6. This supports the idea that Kir2.2 p.Thr140Met conveys susceptibility to SPP and should be included in genetic screening.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:



