Morten Muhlig Nielsen, Paula Tataru, Tobias Madsen, Asger Hobolth, Jakob Skou Pedersen
{"title":"Regmex: a statistical tool for exploring motifs in ranked sequence lists from genomics experiments.","authors":"Morten Muhlig Nielsen, Paula Tataru, Tobias Madsen, Asger Hobolth, Jakob Skou Pedersen","doi":"10.1186/s13015-018-0135-2","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Motif analysis methods have long been central for studying biological function of nucleotide sequences. Functional genomics experiments extend their potential. They typically generate sequence lists ranked by an experimentally acquired functional property such as gene expression or protein binding affinity. Current motif discovery tools suffer from limitations in searching large motif spaces, and thus more complex motifs may not be included. There is thus a need for motif analysis methods that are tailored for analyzing specific complex motifs motivated by biological questions and hypotheses rather than acting as a screen based motif finding tool.</p><p><strong>Methods: </strong>We present Regmex (REGular expression Motif EXplorer), which offers several methods to identify overrepresented motifs in ranked lists of sequences. Regmex uses regular expressions to define motifs or families of motifs and embedded Markov models to calculate exact p-values for motif observations in sequences. Biases in motif distributions across ranked sequence lists are evaluated using random walks, Brownian bridges, or modified rank based statistics. A modular setup and fast analytic <i>p</i> value evaluations make Regmex applicable to diverse and potentially large-scale motif analysis problems.</p><p><strong>Results: </strong>We demonstrate use cases of combined motifs on simulated data and on expression data from micro RNA transfection experiments. We confirm previously obtained results and demonstrate the usability of Regmex to test a specific hypothesis about the relative location of microRNA seed sites and U-rich motifs. We further compare the tool with an existing motif discovery tool and show increased sensitivity.</p><p><strong>Conclusions: </strong>Regmex is a useful and flexible tool to analyze motif hypotheses that relates to large data sets in functional genomics. The method is available as an R package (https://github.com/muhligs/regmex).</p>","PeriodicalId":50823,"journal":{"name":"Algorithms for Molecular Biology","volume":"13 ","pages":"17"},"PeriodicalIF":1.5000,"publicationDate":"2018-12-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s13015-018-0135-2","citationCount":"4","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Algorithms for Molecular Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13015-018-0135-2","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2018/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 4
Abstract
Background: Motif analysis methods have long been central for studying biological function of nucleotide sequences. Functional genomics experiments extend their potential. They typically generate sequence lists ranked by an experimentally acquired functional property such as gene expression or protein binding affinity. Current motif discovery tools suffer from limitations in searching large motif spaces, and thus more complex motifs may not be included. There is thus a need for motif analysis methods that are tailored for analyzing specific complex motifs motivated by biological questions and hypotheses rather than acting as a screen based motif finding tool.
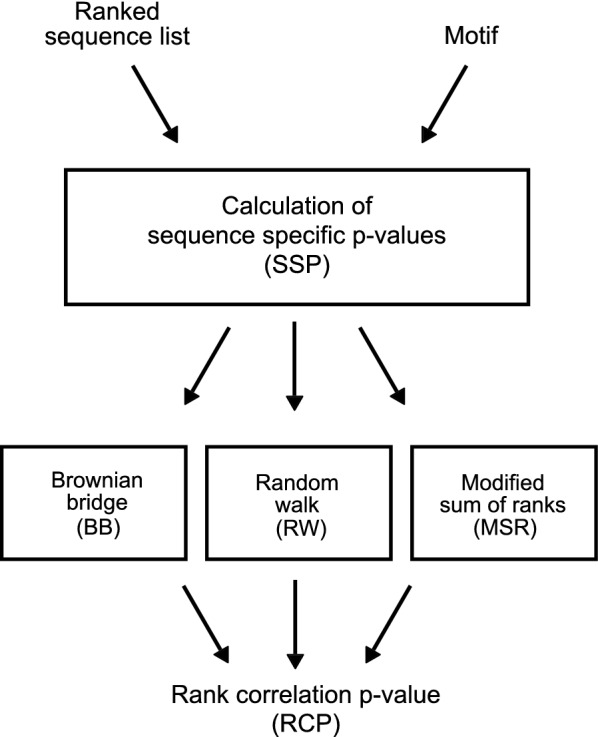
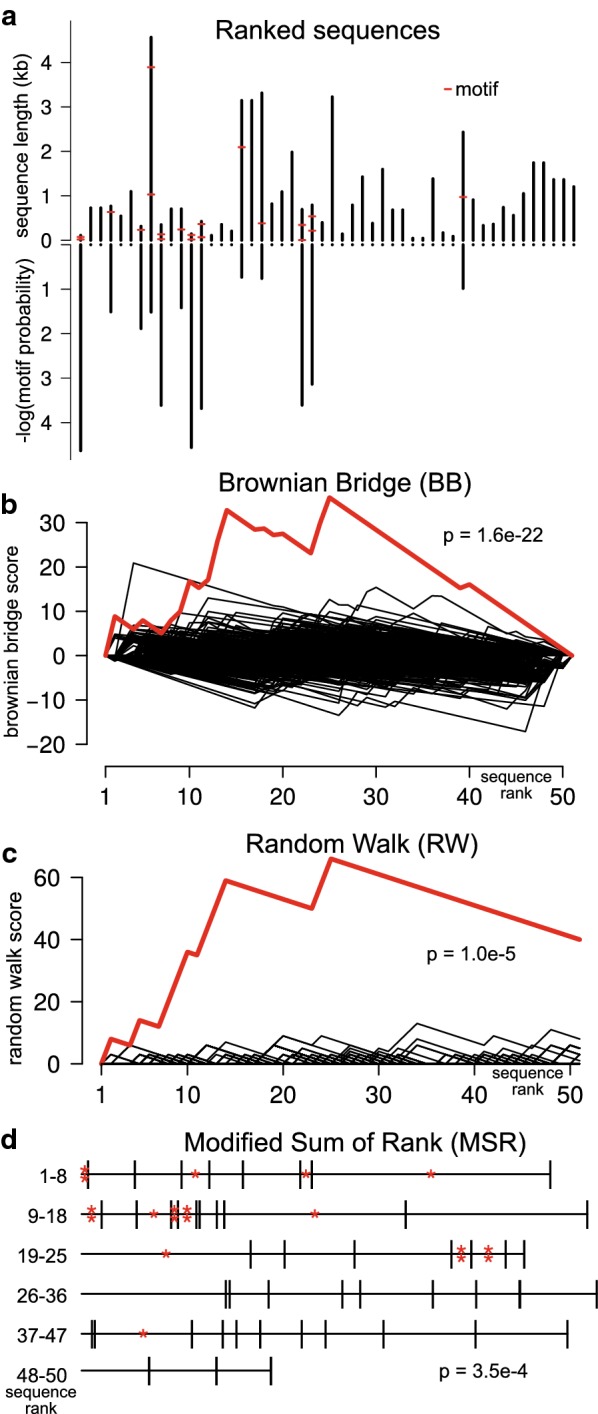
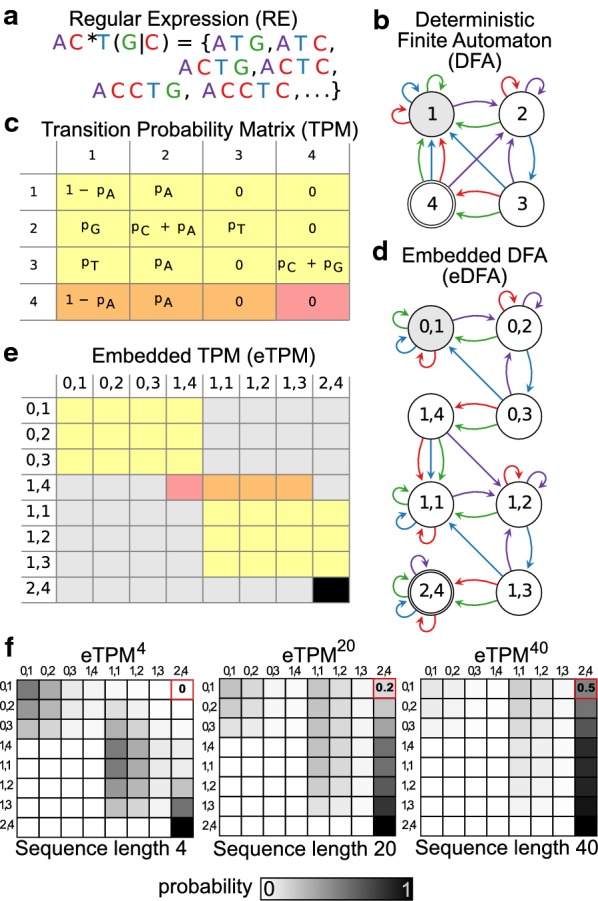
Methods: We present Regmex (REGular expression Motif EXplorer), which offers several methods to identify overrepresented motifs in ranked lists of sequences. Regmex uses regular expressions to define motifs or families of motifs and embedded Markov models to calculate exact p-values for motif observations in sequences. Biases in motif distributions across ranked sequence lists are evaluated using random walks, Brownian bridges, or modified rank based statistics. A modular setup and fast analytic p value evaluations make Regmex applicable to diverse and potentially large-scale motif analysis problems.
Results: We demonstrate use cases of combined motifs on simulated data and on expression data from micro RNA transfection experiments. We confirm previously obtained results and demonstrate the usability of Regmex to test a specific hypothesis about the relative location of microRNA seed sites and U-rich motifs. We further compare the tool with an existing motif discovery tool and show increased sensitivity.
Conclusions: Regmex is a useful and flexible tool to analyze motif hypotheses that relates to large data sets in functional genomics. The method is available as an R package (https://github.com/muhligs/regmex).
期刊介绍:
Algorithms for Molecular Biology publishes articles on novel algorithms for biological sequence and structure analysis, phylogeny reconstruction, and combinatorial algorithms and machine learning.
Areas of interest include but are not limited to: algorithms for RNA and protein structure analysis, gene prediction and genome analysis, comparative sequence analysis and alignment, phylogeny, gene expression, machine learning, and combinatorial algorithms.
Where appropriate, manuscripts should describe applications to real-world data. However, pure algorithm papers are also welcome if future applications to biological data are to be expected, or if they address complexity or approximation issues of novel computational problems in molecular biology. Articles about novel software tools will be considered for publication if they contain some algorithmically interesting aspects.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


