{"title":"Two male siblings with a novel <i>LRBA</i> mutation presenting with different findings of IPEX syndrome.","authors":"Sanem Eren Akarcan, Neslihan Edeer Karaca, Guzide Aksu, Ayca Aykut, Deniz Yilmaz Karapinar, Funda Cetin, Yesim Aydinok, Elif Azarsiz, Eleonora Gambineri, Ozgur Cogulu, Ezgi Ulusoy Severcan, Hudaver Alper, Necil Kutukculer","doi":"10.1099/jmmcr.0.005167","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>LPS-responsive beige-like anchor (LRBA) protein deficiency is a disease of immune dysregulation with autoimmunity affecting various systems.</p><p><strong>Case presentation: </strong>Two male siblings with a novel <i>LRBA</i> mutation had different primary findings at admission: the younger sibling had chronic early-onset diarrhoea and the elder one had autoimmune haemolytic anaemia. During long-term follow-up for IPEX phenotype, both developed hypogammaglobulinaemia, enteropathy and lung involvement. The patients partially responded to immunosuppressive therapies. A homozygous c.2496C>A, p.Cys832Ter (p.C832*) mutation in the <i>LRBA</i> gene causing a premature stop codon was detected. After molecular diagnosis, abatacept, as a target-specific molecule, was used with promising results.</p><p><strong>Conclusion: </strong>LRBA deficiency is a recently defined defect, with variable presentations in different patients; a single, definitive treatment option is thus not yet available.</p>","PeriodicalId":73559,"journal":{"name":"JMM case reports","volume":"5 10","pages":"e005167"},"PeriodicalIF":0.0000,"publicationDate":"2018-10-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1099/jmmcr.0.005167","citationCount":"16","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"JMM case reports","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1099/jmmcr.0.005167","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2018/10/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 16
Abstract
Introduction: LPS-responsive beige-like anchor (LRBA) protein deficiency is a disease of immune dysregulation with autoimmunity affecting various systems.
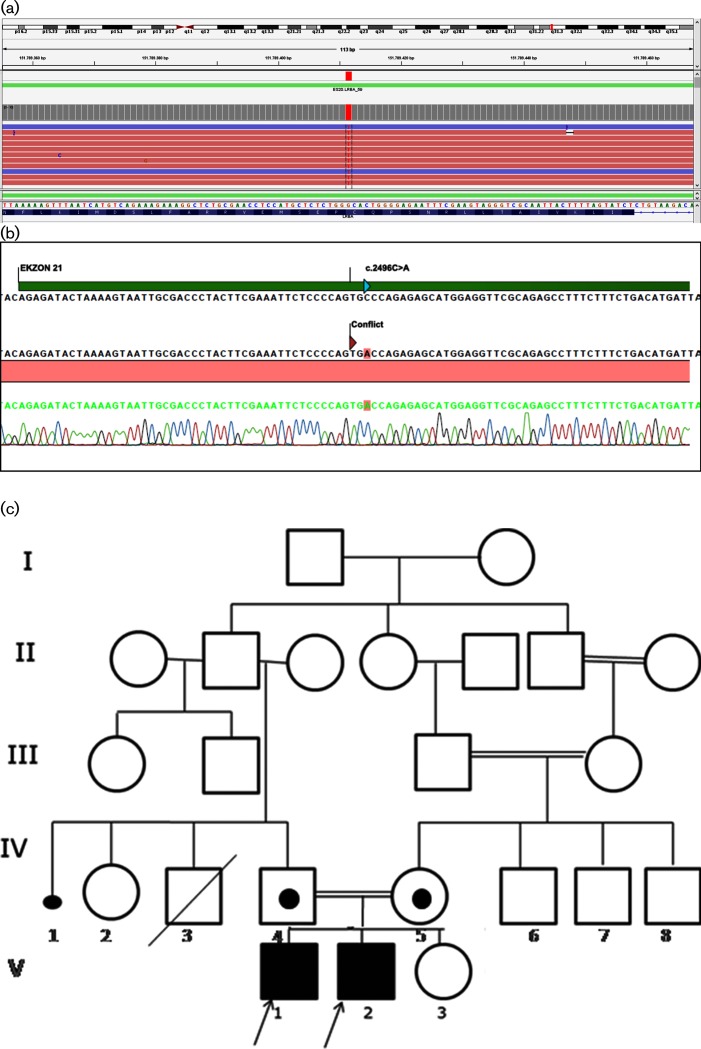
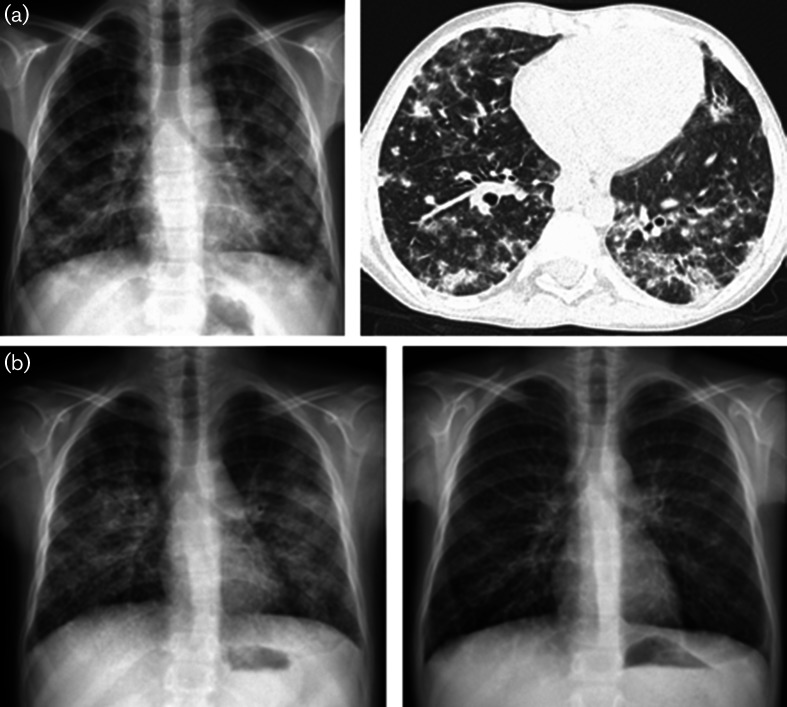
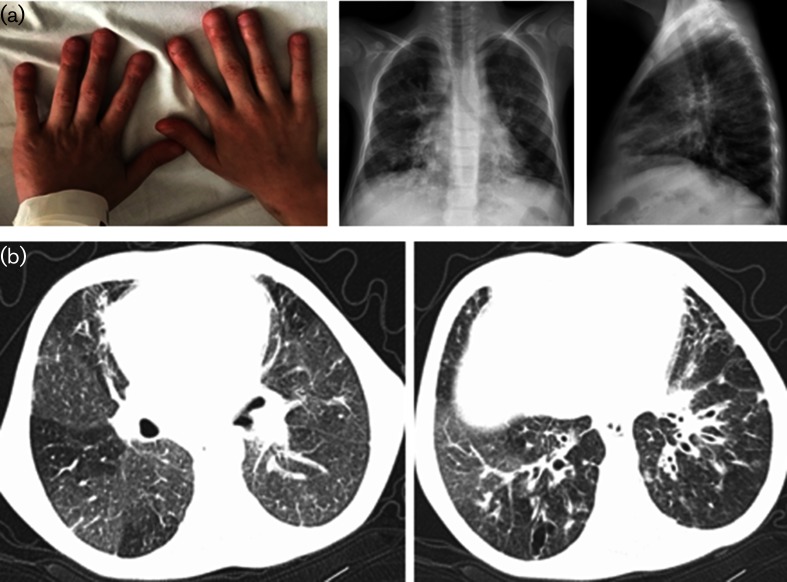
Case presentation: Two male siblings with a novel LRBA mutation had different primary findings at admission: the younger sibling had chronic early-onset diarrhoea and the elder one had autoimmune haemolytic anaemia. During long-term follow-up for IPEX phenotype, both developed hypogammaglobulinaemia, enteropathy and lung involvement. The patients partially responded to immunosuppressive therapies. A homozygous c.2496C>A, p.Cys832Ter (p.C832*) mutation in the LRBA gene causing a premature stop codon was detected. After molecular diagnosis, abatacept, as a target-specific molecule, was used with promising results.
Conclusion: LRBA deficiency is a recently defined defect, with variable presentations in different patients; a single, definitive treatment option is thus not yet available.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


