Justin S Antony, Ngadhnjim Latifi, A K M Ashiqul Haque, Andrés Lamsfus-Calle, Alberto Daniel-Moreno, Sebastian Graeter, Praveen Baskaran, Petra Weinmann, Markus Mezger, Rupert Handgretinger, Michael S D Kormann
{"title":"Gene correction of HBB mutations in CD34<sup>+</sup> hematopoietic stem cells using Cas9 mRNA and ssODN donors.","authors":"Justin S Antony, Ngadhnjim Latifi, A K M Ashiqul Haque, Andrés Lamsfus-Calle, Alberto Daniel-Moreno, Sebastian Graeter, Praveen Baskaran, Petra Weinmann, Markus Mezger, Rupert Handgretinger, Michael S D Kormann","doi":"10.1186/s40348-018-0086-1","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>β-Thalassemia is an inherited hematological disorder caused by mutations in the human hemoglobin beta (HBB) gene that reduce or abrogate β-globin expression. Although lentiviral-mediated expression of β-globin and autologous transplantation is a promising therapeutic approach, the risk of insertional mutagenesis or low transgene expression is apparent. However, targeted gene correction of HBB mutations with programmable nucleases such as CRISPR/Cas9, TALENs, and ZFNs with non-viral repair templates ensures a higher safety profile and endogenous expression control.</p><p><strong>Methods: </strong>We have compared three different gene-editing tools (CRISPR/Cas9, TALENs, and ZFNs) for their targeting efficiency of the HBB gene locus. As a proof of concept, we studied the personalized gene-correction therapy for a common β-thalassemia splicing variant HBB<sup>IVS1-110</sup> using Cas9 mRNA and several optimally designed single-stranded oligonucleotide (ssODN) donors in K562 and CD34<sup>+</sup> hematopoietic stem cells (HSCs).</p><p><strong>Results: </strong>Our results exhibited that indel frequency of CRISPR/Cas9 was superior to TALENs and ZFNs (P < 0.0001). Our designed sgRNA targeting the site of HBB<sup>IVS1-110</sup> mutation showed indels in both K562 cells (up to 77%) and CD34<sup>+</sup> hematopoietic stem cells-HSCs (up to 87%). The absolute quantification by next-generation sequencing showed that up to 8% site-specific insertion of the NheI tag was achieved using Cas9 mRNA and a chemically modified ssODN in CD34<sup>+</sup> HSCs.</p><p><strong>Conclusion: </strong>Our approach provides guidance on non-viral gene correction in CD34<sup>+</sup> HSCs using Cas9 mRNA and chemically modified ssODN. However, further optimization is needed to increase the homology directed repair (HDR) to attain a real clinical benefit for β-thalassemia.</p>","PeriodicalId":74215,"journal":{"name":"Molecular and cellular pediatrics","volume":"5 1","pages":"9"},"PeriodicalIF":2.4000,"publicationDate":"2018-11-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s40348-018-0086-1","citationCount":"43","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular and cellular pediatrics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s40348-018-0086-1","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"PEDIATRICS","Score":null,"Total":0}
引用次数: 43
Abstract
Background: β-Thalassemia is an inherited hematological disorder caused by mutations in the human hemoglobin beta (HBB) gene that reduce or abrogate β-globin expression. Although lentiviral-mediated expression of β-globin and autologous transplantation is a promising therapeutic approach, the risk of insertional mutagenesis or low transgene expression is apparent. However, targeted gene correction of HBB mutations with programmable nucleases such as CRISPR/Cas9, TALENs, and ZFNs with non-viral repair templates ensures a higher safety profile and endogenous expression control.
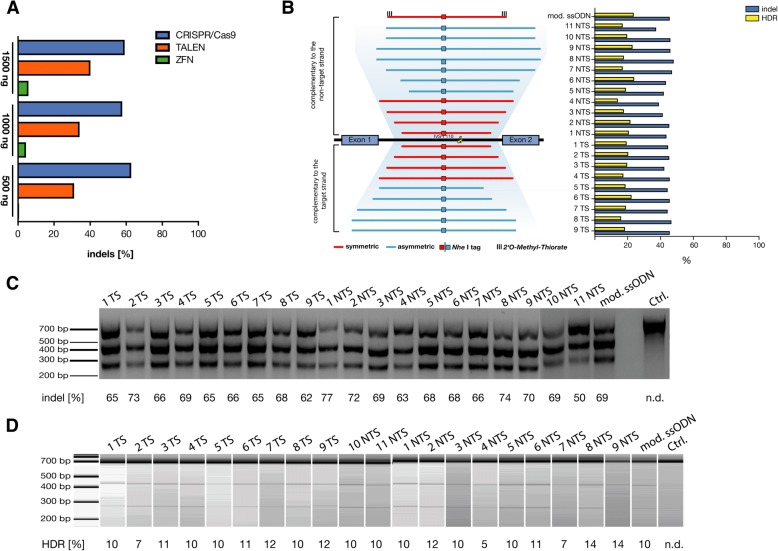
Methods: We have compared three different gene-editing tools (CRISPR/Cas9, TALENs, and ZFNs) for their targeting efficiency of the HBB gene locus. As a proof of concept, we studied the personalized gene-correction therapy for a common β-thalassemia splicing variant HBBIVS1-110 using Cas9 mRNA and several optimally designed single-stranded oligonucleotide (ssODN) donors in K562 and CD34+ hematopoietic stem cells (HSCs).
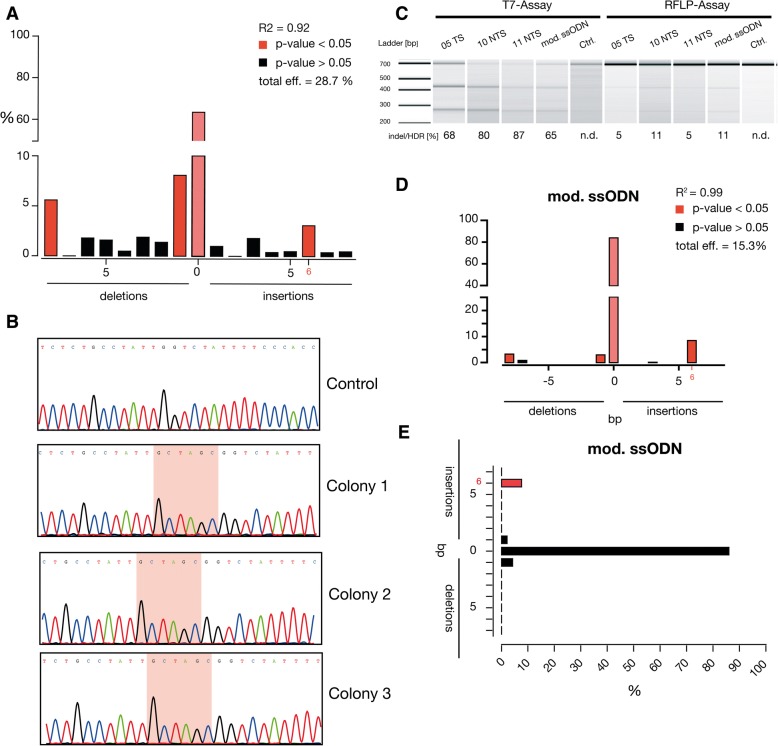
Results: Our results exhibited that indel frequency of CRISPR/Cas9 was superior to TALENs and ZFNs (P < 0.0001). Our designed sgRNA targeting the site of HBBIVS1-110 mutation showed indels in both K562 cells (up to 77%) and CD34+ hematopoietic stem cells-HSCs (up to 87%). The absolute quantification by next-generation sequencing showed that up to 8% site-specific insertion of the NheI tag was achieved using Cas9 mRNA and a chemically modified ssODN in CD34+ HSCs.
Conclusion: Our approach provides guidance on non-viral gene correction in CD34+ HSCs using Cas9 mRNA and chemically modified ssODN. However, further optimization is needed to increase the homology directed repair (HDR) to attain a real clinical benefit for β-thalassemia.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


