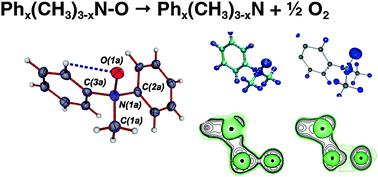
{"title":"Bonding situation and N–O-bond strengths in amine-N-oxides—a combined experimental and theoretical study†","authors":"Andrey Yu. Rogachev and Peter Burger","doi":"10.1039/C2CP22341D","DOIUrl":null,"url":null,"abstract":"<p >The bonding situation and energetics of the N–O bond in a series of amine-<em>N</em>-oxides, Ph<small><sub><em>x</em></sub></small>(CH<small><sub>3</sub></small>)<small><sub>3?<em>x</em></sub></small>N–O, where <em>x</em> = 0–3, were analyzed experimentally and theoretically. There is a notable nearly linear decrease of the N–O bond dissociation energies (BDEs) for this series with an increasing number of <annref>phenyl</annref> groups<em>x</em>. This was investigated experimentally by X-ray high angle multipole refinement techniques in combination with subsequent topological analysis of the electron density for the representative (CH<small><sub>3</sub></small>)<small><sub>2</sub></small>PhN–O, <strong>2</strong>, and complementary theoretical calculations at the DFT and multireference CASSCF and MR-perturbation theory (MCQDPT2) levels. Both the theoretical and experimental results unambiguously revealed a polar covalent σ-bond for the N–O bond with an essentially identical bonding situation for all amine-<em>N</em>-oxides studied. This apparent disparity between the bonding situation and the trend of BDEs is attributed to the large differences of the relaxation energies of the corresponding amines Ph<small><sub><em>x</em></sub></small>(CH<small><sub>3</sub></small>)<small><sub>3?<em>x</em></sub></small>N, (<em>x</em> = 0–3), respectively, the required preparation energies (Δ<em>E</em><small><sub>prep</sub></small>) for the reverse N–O bond forming process. The detailed theoretical analysis of the <annref>amines</annref> allowed us to trace the trend of larger values of Δ<em>E</em><small><sub>prep</sub></small> for a higher number of <annref>phenyl</annref> groups<em>x</em> to an increase of <em>n</em>(N) → π*(C–C) delocalization interactions.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 6","pages":" 1985-2000"},"PeriodicalIF":2.9000,"publicationDate":"2012-01-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1039/C2CP22341D","citationCount":"15","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2012/cp/c2cp22341d","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 15
Abstract
The bonding situation and energetics of the N–O bond in a series of amine-N-oxides, Phx(CH3)3?xN–O, where x = 0–3, were analyzed experimentally and theoretically. There is a notable nearly linear decrease of the N–O bond dissociation energies (BDEs) for this series with an increasing number of phenyl groupsx. This was investigated experimentally by X-ray high angle multipole refinement techniques in combination with subsequent topological analysis of the electron density for the representative (CH3)2PhN–O, 2, and complementary theoretical calculations at the DFT and multireference CASSCF and MR-perturbation theory (MCQDPT2) levels. Both the theoretical and experimental results unambiguously revealed a polar covalent σ-bond for the N–O bond with an essentially identical bonding situation for all amine-N-oxides studied. This apparent disparity between the bonding situation and the trend of BDEs is attributed to the large differences of the relaxation energies of the corresponding amines Phx(CH3)3?xN, (x = 0–3), respectively, the required preparation energies (ΔEprep) for the reverse N–O bond forming process. The detailed theoretical analysis of the amines allowed us to trace the trend of larger values of ΔEprep for a higher number of phenyl groupsx to an increase of n(N) → π*(C–C) delocalization interactions.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


