Integration of Genome-Wide DNA Methylation and Transcription Uncovered Aberrant Methylation-Regulated Genes and Pathways in the Peripheral Blood Mononuclear Cells of Systemic Sclerosis.
Honglin Zhu, Chengsong Zhu, Wentao Mi, Tao Chen, Hongjun Zhao, Xiaoxia Zuo, Hui Luo, Quan-Zhen Li
{"title":"Integration of Genome-Wide DNA Methylation and Transcription Uncovered Aberrant Methylation-Regulated Genes and Pathways in the Peripheral Blood Mononuclear Cells of Systemic Sclerosis.","authors":"Honglin Zhu, Chengsong Zhu, Wentao Mi, Tao Chen, Hongjun Zhao, Xiaoxia Zuo, Hui Luo, Quan-Zhen Li","doi":"10.1155/2018/7342472","DOIUrl":null,"url":null,"abstract":"<p><p><i>Objective.</i> Systemic sclerosis (SSc) is a systemic connective tissue disease of unknown etiology. Aberrant gene expression and epigenetic modifications in circulating immune cells have been implicated in the pathogenesis of SSc. This study is to delineate the interaction network between gene transcription and DNA methylation in PBMC of SSc patients and to identify methylation-regulated genes which are involved in the pathogenesis of SSc. <i>Methods.</i> Genome-wide mRNA transcription and global DNA methylation analysis were performed on PBMC from 18 SSc patients and 19 matched normal controls (NC) using Illumina BeadChips. Differentially expressed genes (DEGs) and differentially methylated positions (DMPs) were integrative analyzed to identify methylation-regulated genes and associated molecular pathways<i>. Results.</i> Transcriptome analysis distinguished 453 DEGs (269 up- and 184 downregulated) in SSc from NC. Global DNA methylation analysis identified 925 DMPs located on 618 genes. Integration of the two lists revealed only 20 DEGs which harbor inversely correlated DMPs, including 12 upregulated (ELANE, CTSG, LTBR, C3AR1, CSTA, SPI1, ODF3B, SAMD4A, PLAUR, NFE2, ZYX, and CTSZ) and eight downregulated genes (RUNX3, PRF1, PRKCH, PAG1, RASSF5, FYN, CXCR6, and F2R). These potential methylation-regulated DEGs (MeDEGs) are enriched in the pathways related to immune cell migration, proliferation, activation, and inflammation activities. Using a machine learning algorism, we identified six out of the 20 MeDEGs, including F2R, CXCR6, FYN, LTBR, CTSG, and ELANE, which distinguished SSc from NC with 100% accuracy. Four genes (F2R, FYN, PAG1, and PRKCH) differentially expressed in SSc with interstitial lung disease (ILD) compared to SSc without ILD. <i>Conclusion.</i> The identified MeDEGs may represent novel candidate factors which lead to the abnormal activation of immune regulatory pathways in the pathogenesis of SSc. They may also be used as diagnostic biomarkers for SSc and clinical complications.</p>","PeriodicalId":51715,"journal":{"name":"International Journal of Rheumatology","volume":"2018 ","pages":"7342472"},"PeriodicalIF":2.3000,"publicationDate":"2018-09-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1155/2018/7342472","citationCount":"21","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Rheumatology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2018/7342472","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2018/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"RHEUMATOLOGY","Score":null,"Total":0}
引用次数: 21
Abstract
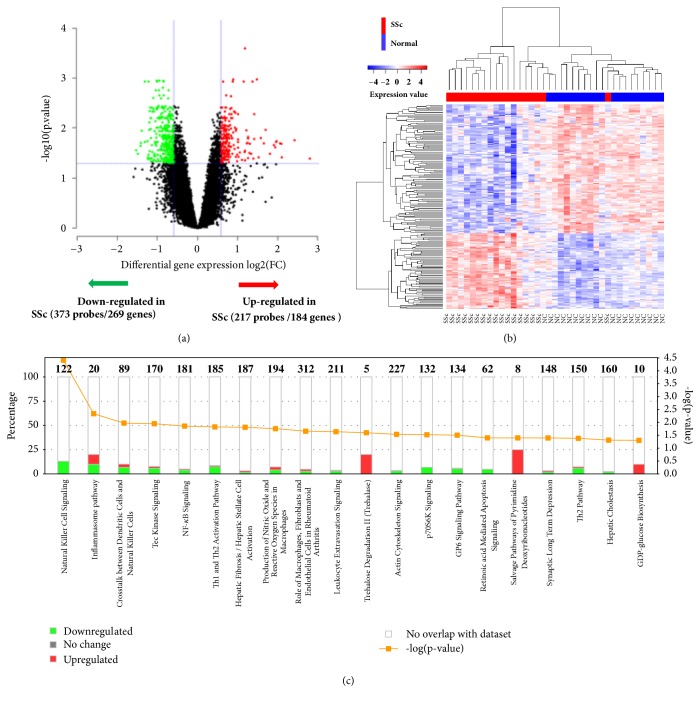
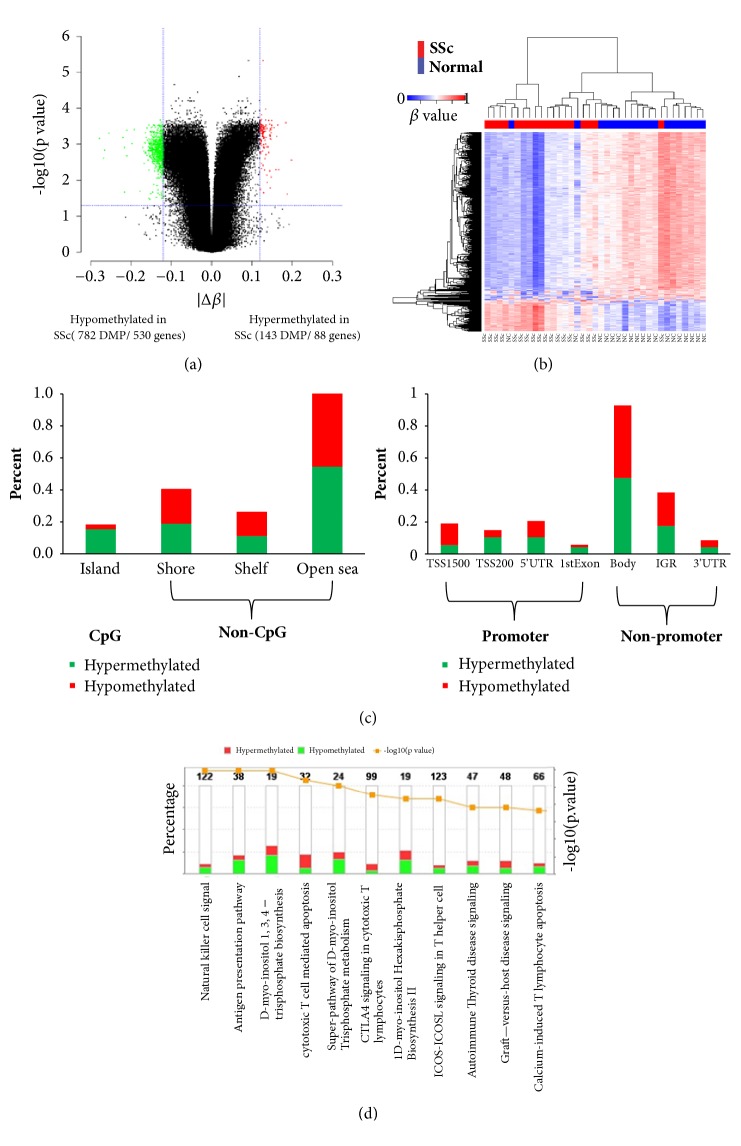
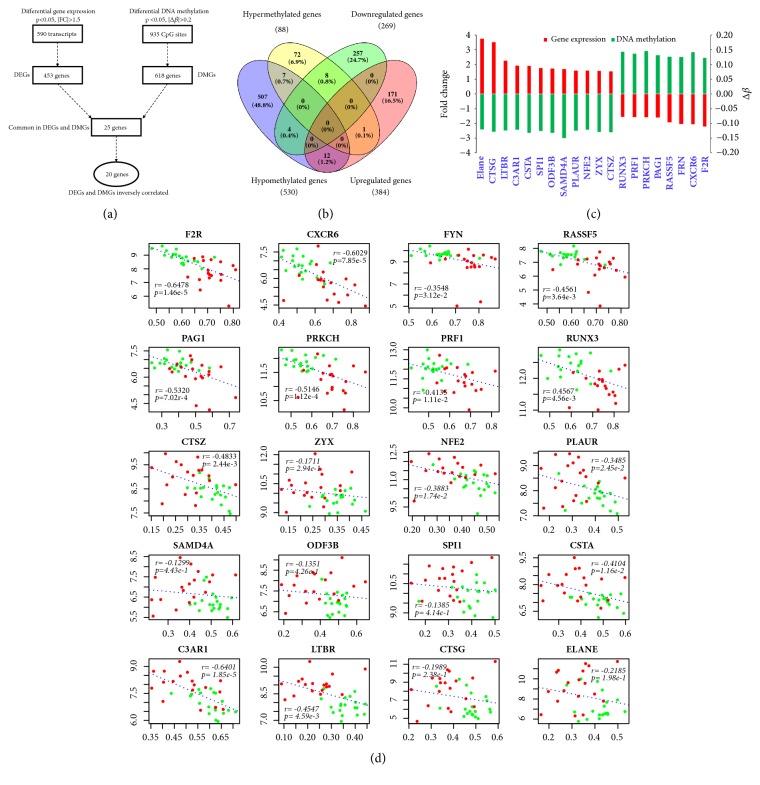
Objective. Systemic sclerosis (SSc) is a systemic connective tissue disease of unknown etiology. Aberrant gene expression and epigenetic modifications in circulating immune cells have been implicated in the pathogenesis of SSc. This study is to delineate the interaction network between gene transcription and DNA methylation in PBMC of SSc patients and to identify methylation-regulated genes which are involved in the pathogenesis of SSc. Methods. Genome-wide mRNA transcription and global DNA methylation analysis were performed on PBMC from 18 SSc patients and 19 matched normal controls (NC) using Illumina BeadChips. Differentially expressed genes (DEGs) and differentially methylated positions (DMPs) were integrative analyzed to identify methylation-regulated genes and associated molecular pathways. Results. Transcriptome analysis distinguished 453 DEGs (269 up- and 184 downregulated) in SSc from NC. Global DNA methylation analysis identified 925 DMPs located on 618 genes. Integration of the two lists revealed only 20 DEGs which harbor inversely correlated DMPs, including 12 upregulated (ELANE, CTSG, LTBR, C3AR1, CSTA, SPI1, ODF3B, SAMD4A, PLAUR, NFE2, ZYX, and CTSZ) and eight downregulated genes (RUNX3, PRF1, PRKCH, PAG1, RASSF5, FYN, CXCR6, and F2R). These potential methylation-regulated DEGs (MeDEGs) are enriched in the pathways related to immune cell migration, proliferation, activation, and inflammation activities. Using a machine learning algorism, we identified six out of the 20 MeDEGs, including F2R, CXCR6, FYN, LTBR, CTSG, and ELANE, which distinguished SSc from NC with 100% accuracy. Four genes (F2R, FYN, PAG1, and PRKCH) differentially expressed in SSc with interstitial lung disease (ILD) compared to SSc without ILD. Conclusion. The identified MeDEGs may represent novel candidate factors which lead to the abnormal activation of immune regulatory pathways in the pathogenesis of SSc. They may also be used as diagnostic biomarkers for SSc and clinical complications.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


