Edith Schussler, Rita V Linkner, Jacob Levitt, Lakshmi Mehta, John A Martignetti, Kimihiko Oishi
{"title":"Protein-losing enteropathy and joint contractures caused by a novel homozygous ANTXR2 mutation.","authors":"Edith Schussler, Rita V Linkner, Jacob Levitt, Lakshmi Mehta, John A Martignetti, Kimihiko Oishi","doi":"10.2147/AGG.S159077","DOIUrl":null,"url":null,"abstract":"<p><p>Infantile systemic hyalinosis (ISH) is a rare autosomal recessive disorder and an allelic form of hyaline fibromatosis syndrome that is caused by mutations in the <i>ANTRX2</i> gene encoding the transmembrane anthrax toxin receptor 2. Its main features include characteristic skin lesions, joint contractures, persistent diarrhea, and failure to thrive due to accumulation of hyaline material in multiple organs. The resulting severe malnutrition can cause death in early infancy. Because of its rarity and high fatality rate, timely diagnosis is difficult and ISH may be underdiagnosed. In this report, we describe a 10-month-old male with severe protein-losing enteropathy, skin lesions, and painful joint contractures, diagnosed with ISH based on skin his-topathology and identification of a novel homozygous <i>ANTRX2</i> mutation, c.1127_1128delTG (p.V376Gfs*14). While its clinical outcome is poor without curative treatment, establishing a diagnosis of ISH starting from clinical suspicion to molecular analysis is important for appropriate medical management and for risk and carrier assessment of family members.</p>","PeriodicalId":89652,"journal":{"name":"Advances in genomics and genetics","volume":"8 ","pages":"17-21"},"PeriodicalIF":0.0000,"publicationDate":"2018-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.2147/AGG.S159077","citationCount":"6","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Advances in genomics and genetics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.2147/AGG.S159077","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2018/6/27 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 6
Abstract
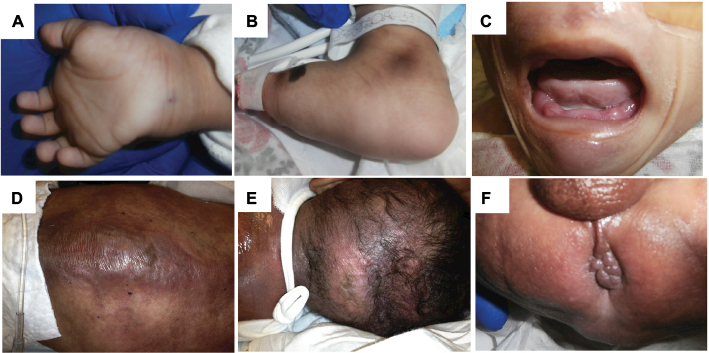
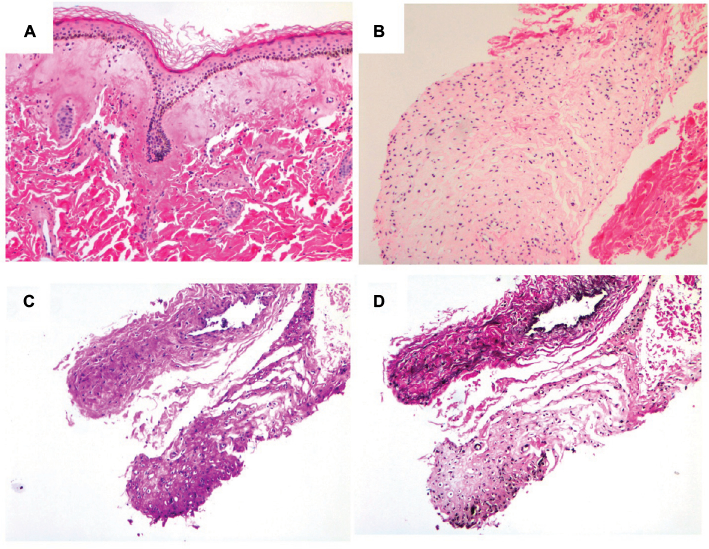
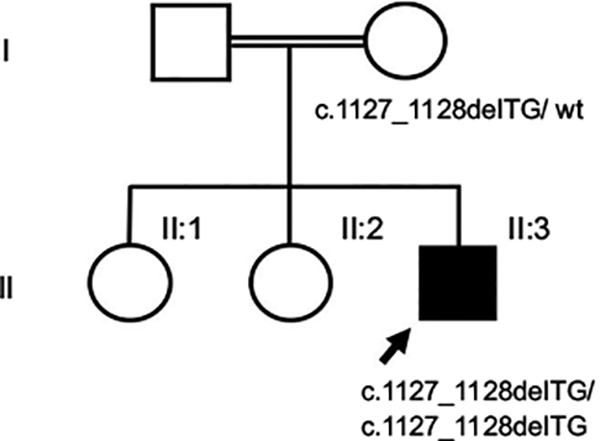
Infantile systemic hyalinosis (ISH) is a rare autosomal recessive disorder and an allelic form of hyaline fibromatosis syndrome that is caused by mutations in the ANTRX2 gene encoding the transmembrane anthrax toxin receptor 2. Its main features include characteristic skin lesions, joint contractures, persistent diarrhea, and failure to thrive due to accumulation of hyaline material in multiple organs. The resulting severe malnutrition can cause death in early infancy. Because of its rarity and high fatality rate, timely diagnosis is difficult and ISH may be underdiagnosed. In this report, we describe a 10-month-old male with severe protein-losing enteropathy, skin lesions, and painful joint contractures, diagnosed with ISH based on skin his-topathology and identification of a novel homozygous ANTRX2 mutation, c.1127_1128delTG (p.V376Gfs*14). While its clinical outcome is poor without curative treatment, establishing a diagnosis of ISH starting from clinical suspicion to molecular analysis is important for appropriate medical management and for risk and carrier assessment of family members.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


