Afnan A Alsultan, Rachel Waller, Paul R Heath, Janine Kirby
{"title":"The genetics of amyotrophic lateral sclerosis: current insights.","authors":"Afnan A Alsultan, Rachel Waller, Paul R Heath, Janine Kirby","doi":"10.2147/DNND.S84956","DOIUrl":null,"url":null,"abstract":"<p><p>Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disorder that results in loss of the upper and lower motor neurons from motor cortex, brainstem, and spinal cord. While the majority of cases are sporadic, approximately 10% show familial inheritance. ALS is usually inherited in an autosomal dominant manner, although autosomal recessive and X-linked inheritance do occur. To date, 24 of the genes at 26 loci have been identified; these include loci linked to ALS and to frontotemporal dementia-ALS, where family pedigrees contain individuals with frontotemporal dementia with/without ALS. The most commonly established genetic causes of familial ALS (FALS) to date are the presence of a hexanucleotide repeat expansion in the <i>C9ORF72</i> gene (39.3% FALS) and mutation of <i>SOD1</i>, <i>TARDBP</i>, and <i>FUS</i>, with frequencies of 12%-23.5%, 5%, and 4.1%, respectively. However, with the increasing use of next-generation sequencing of small family pedigrees, this has led to an increasing number of genes being associated with ALS. This review provides a comprehensive review on the genetics of ALS and an update of the pathogenic mechanisms associated with these genes. Commonly implicated pathways have been established, including RNA processing, the protein degradation pathways of autophagy and ubiquitin-proteasome system, as well as protein trafficking and cytoskeletal function. Elucidating the role genetics plays in both FALS and sporadic ALS is essential for understanding the subsequent cellular dysregulation that leads to motor neuron loss, in order to develop future effective therapeutic strategies.</p>","PeriodicalId":11147,"journal":{"name":"Degenerative Neurological and Neuromuscular Disease","volume":"6 ","pages":"49-64"},"PeriodicalIF":0.0000,"publicationDate":"2016-05-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/36/14/dnnd-6-049.PMC6053097.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Degenerative Neurological and Neuromuscular Disease","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.2147/DNND.S84956","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2016/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
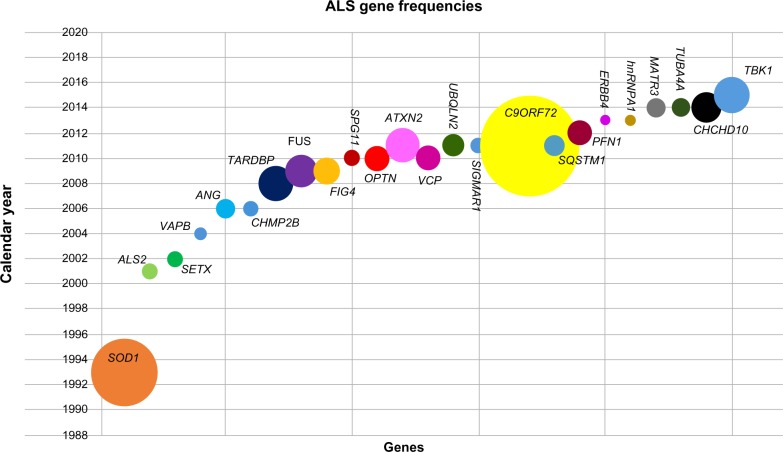
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disorder that results in loss of the upper and lower motor neurons from motor cortex, brainstem, and spinal cord. While the majority of cases are sporadic, approximately 10% show familial inheritance. ALS is usually inherited in an autosomal dominant manner, although autosomal recessive and X-linked inheritance do occur. To date, 24 of the genes at 26 loci have been identified; these include loci linked to ALS and to frontotemporal dementia-ALS, where family pedigrees contain individuals with frontotemporal dementia with/without ALS. The most commonly established genetic causes of familial ALS (FALS) to date are the presence of a hexanucleotide repeat expansion in the C9ORF72 gene (39.3% FALS) and mutation of SOD1, TARDBP, and FUS, with frequencies of 12%-23.5%, 5%, and 4.1%, respectively. However, with the increasing use of next-generation sequencing of small family pedigrees, this has led to an increasing number of genes being associated with ALS. This review provides a comprehensive review on the genetics of ALS and an update of the pathogenic mechanisms associated with these genes. Commonly implicated pathways have been established, including RNA processing, the protein degradation pathways of autophagy and ubiquitin-proteasome system, as well as protein trafficking and cytoskeletal function. Elucidating the role genetics plays in both FALS and sporadic ALS is essential for understanding the subsequent cellular dysregulation that leads to motor neuron loss, in order to develop future effective therapeutic strategies.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


