{"title":"Split-inducing indels in phylogenomic analysis.","authors":"Alexander Donath, Peter F Stadler","doi":"10.1186/s13015-018-0130-7","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Most phylogenetic studies using molecular data treat gaps in multiple sequence alignments as missing data or even completely exclude alignment columns that contain gaps.</p><p><strong>Results: </strong>Here we show that gap patterns in large-scale, genome-wide alignments are themselves phylogenetically informative and can be used to infer reliable phylogenies provided the gap data are properly filtered to reduce noise introduced by the alignment method. We introduce here the notion of split-inducing indels (<i>splids</i>) that define an approximate bipartition of the taxon set. We show both in simulated data and in case studies on real-life data that <i>splids</i> can be efficiently extracted from phylogenomic data sets.</p><p><strong>Conclusions: </strong>Suitably processed gap patterns extracted from genome-wide alignment provide a surprisingly clear phylogenetic signal and an allow the inference of accurate phylogenetic trees.</p>","PeriodicalId":50823,"journal":{"name":"Algorithms for Molecular Biology","volume":"13 ","pages":"12"},"PeriodicalIF":1.7000,"publicationDate":"2018-07-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s13015-018-0130-7","citationCount":"11","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Algorithms for Molecular Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13015-018-0130-7","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2018/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 11
Abstract
Background: Most phylogenetic studies using molecular data treat gaps in multiple sequence alignments as missing data or even completely exclude alignment columns that contain gaps.
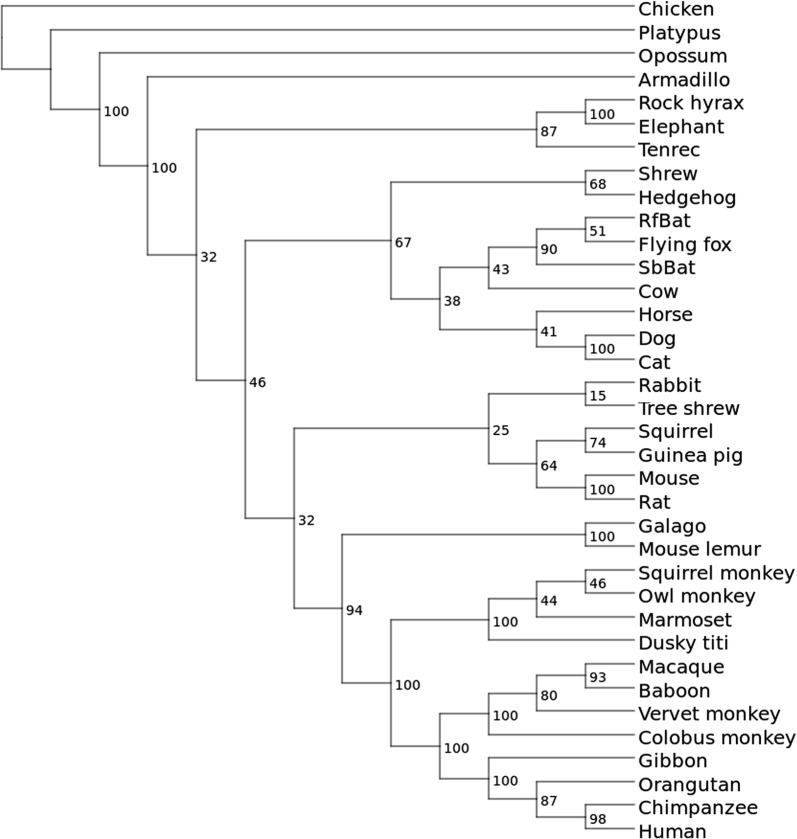
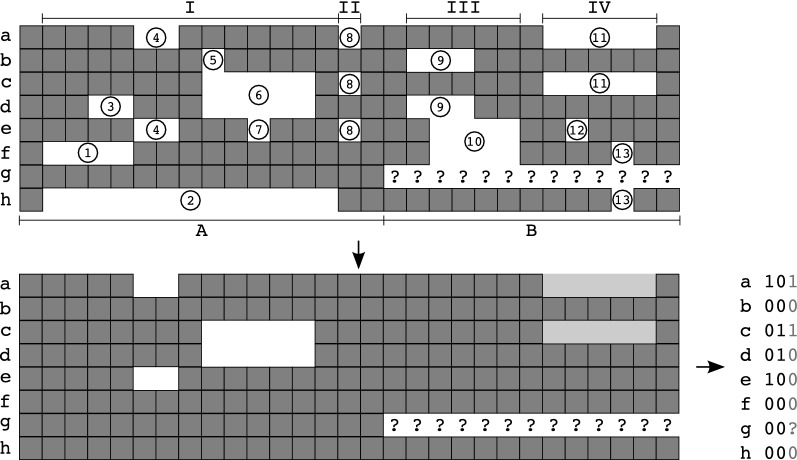
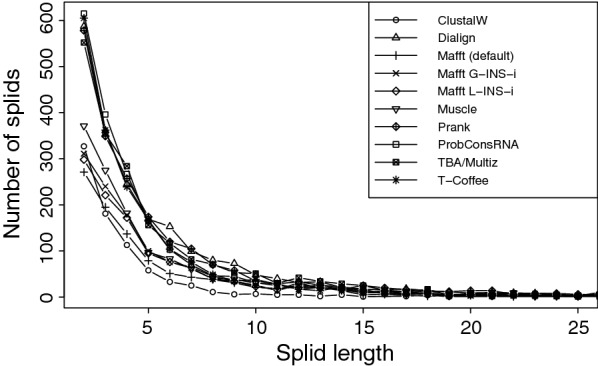
Results: Here we show that gap patterns in large-scale, genome-wide alignments are themselves phylogenetically informative and can be used to infer reliable phylogenies provided the gap data are properly filtered to reduce noise introduced by the alignment method. We introduce here the notion of split-inducing indels (splids) that define an approximate bipartition of the taxon set. We show both in simulated data and in case studies on real-life data that splids can be efficiently extracted from phylogenomic data sets.
Conclusions: Suitably processed gap patterns extracted from genome-wide alignment provide a surprisingly clear phylogenetic signal and an allow the inference of accurate phylogenetic trees.
期刊介绍:
Algorithms for Molecular Biology publishes articles on novel algorithms for biological sequence and structure analysis, phylogeny reconstruction, and combinatorial algorithms and machine learning.
Areas of interest include but are not limited to: algorithms for RNA and protein structure analysis, gene prediction and genome analysis, comparative sequence analysis and alignment, phylogeny, gene expression, machine learning, and combinatorial algorithms.
Where appropriate, manuscripts should describe applications to real-world data. However, pure algorithm papers are also welcome if future applications to biological data are to be expected, or if they address complexity or approximation issues of novel computational problems in molecular biology. Articles about novel software tools will be considered for publication if they contain some algorithmically interesting aspects.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


