{"title":"Physicochemical Prediction of Metabolite Fragmentation in Tandem Mass Spectrometry.","authors":"Wataru Tanaka, Masanori Arita","doi":"10.5702/massspectrometry.A0066","DOIUrl":null,"url":null,"abstract":"<p><p>Current bottleneck of comprehensive non-target metabolite identification is insufficient spectral library. Many research groups have tried to build a theoretical product ion spectral library independent of measurement condition or settings, but mechanisms of metabolite fragmentation are not fully clarified. To achieve the mechanistic prediction of metabolite fragmentation which covers a wide range of metabolites, we will discuss utilization of physicochemical calculation. We introduce bonding patterns, which include two bound atoms and chemical groups adjacent to the bond. Cleavage of each bonding pattern is simulated and its activation energy is precisely calculated with quantum chemistry and assigned on metabolites. By tracing low-energy bond cleavages, fragmentation of a dipeptide molecule is successfully predicted. Prediction on another metabolite requires some additional features to fully reproduce its experimentally observed product ions. Physicochemical calculation shows its promising ability to predict fragmentation pathways only from metabolite structures, while required improvements suggested by comparison between our prediction and standard spectra stored in database are also discussed. Moreover, to construct a prediction strategy which withstands the vast metabolite space, we have to build a comprehensive list of bonding patterns and their activation energy. As theoretically possible bonding patterns are huge in number, proper simplification of the patterns must be implemented. We will discuss how to achieve it in addition to the prediction results.</p>","PeriodicalId":18243,"journal":{"name":"Mass spectrometry","volume":"7 1","pages":"A0066"},"PeriodicalIF":0.0000,"publicationDate":"2018-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.5702/massspectrometry.A0066","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Mass spectrometry","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.5702/massspectrometry.A0066","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2018/6/14 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"Physics and Astronomy","Score":null,"Total":0}
引用次数: 0
Abstract
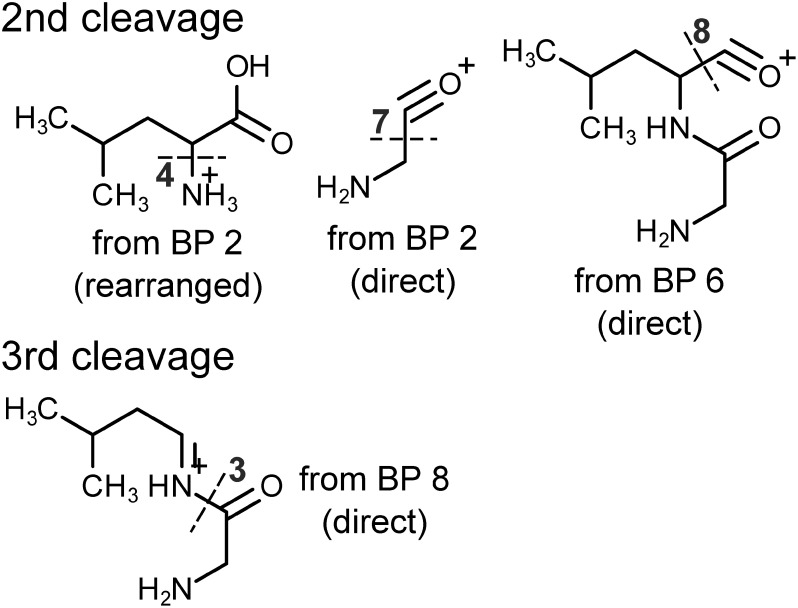
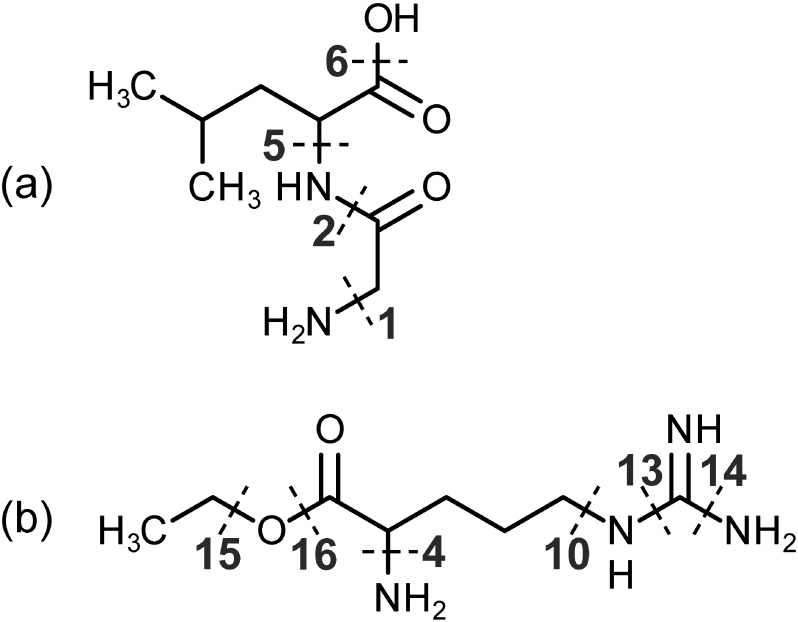
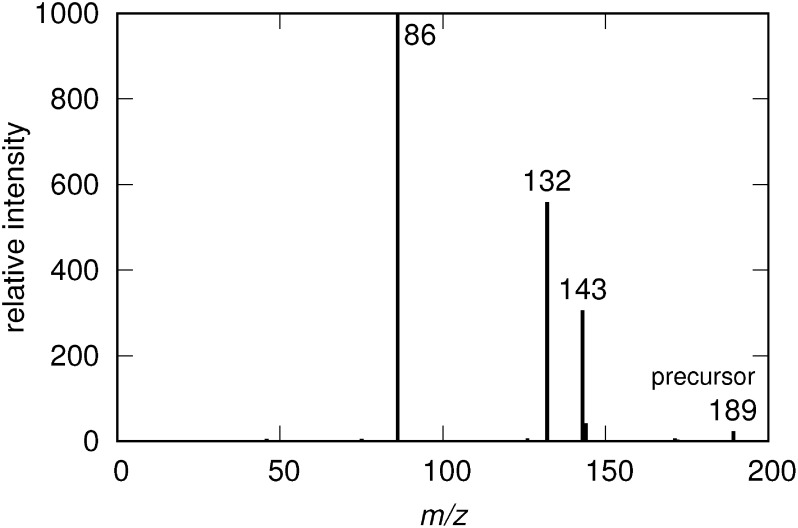
Current bottleneck of comprehensive non-target metabolite identification is insufficient spectral library. Many research groups have tried to build a theoretical product ion spectral library independent of measurement condition or settings, but mechanisms of metabolite fragmentation are not fully clarified. To achieve the mechanistic prediction of metabolite fragmentation which covers a wide range of metabolites, we will discuss utilization of physicochemical calculation. We introduce bonding patterns, which include two bound atoms and chemical groups adjacent to the bond. Cleavage of each bonding pattern is simulated and its activation energy is precisely calculated with quantum chemistry and assigned on metabolites. By tracing low-energy bond cleavages, fragmentation of a dipeptide molecule is successfully predicted. Prediction on another metabolite requires some additional features to fully reproduce its experimentally observed product ions. Physicochemical calculation shows its promising ability to predict fragmentation pathways only from metabolite structures, while required improvements suggested by comparison between our prediction and standard spectra stored in database are also discussed. Moreover, to construct a prediction strategy which withstands the vast metabolite space, we have to build a comprehensive list of bonding patterns and their activation energy. As theoretically possible bonding patterns are huge in number, proper simplification of the patterns must be implemented. We will discuss how to achieve it in addition to the prediction results.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


