Massimo Delle Piane, Marta Corno, Roberto Orlando, Roberto Dovesi, Piero Ugliengo
{"title":"Elucidating the fundamental forces in protein crystal formation: the case of crambin.","authors":"Massimo Delle Piane, Marta Corno, Roberto Orlando, Roberto Dovesi, Piero Ugliengo","doi":"10.1039/c5sc03447g","DOIUrl":null,"url":null,"abstract":"<p><p>Molecular simulations of proteins have been usually accomplished through empirical or semi-empirical potentials, due to the large size and inherent complexity of these biological systems. On the other hand, a theoretical description of proteins based on quantum-mechanical methods would however provide an unbiased characterization of their electronic properties, possibly offering a link between these and the ultimate biological activity. Yet, such approaches have been historically hindered by the large amount of requested computational power. Here we demonstrate the feasibility of periodic all-electron density functional theory calculations in the description of the crystal of the protein crambin (46 aminoacids), which is determined with exceptional structural accuracy. We have employed the hybrid B3LYP functional, coupled to an empirical description of London interactions (D*) to simulate the crambin crystal with an increasing amount of lattice water molecules in the cell (up to 172H<sub>2</sub>O per cell). The agreement with the experiment is good for both protein geometry and protein-water interactions. The energetics was computed to predict crystal formation energies, protein-water and protein-protein interaction energies. We studied the role of dispersion interactions which are crucial for holding the crambin crystal in place. B3LYP-D* electrostatic potential and dipole moment of crambin as well as the electronic charge flow from crambin to the solvating water molecules (0.0015<i>e</i> per H<sub>2</sub>O) have also been predicted. These results proved that quantum-mechanical simulations of small proteins, both free and in their crystalline state, are now feasible in a reasonable amount of time, by programs capable of exploiting high performance computing architectures, allowing the study of protein properties not easily amenable through classical force fields.</p>","PeriodicalId":9909,"journal":{"name":"Chemical Science","volume":"7 2","pages":"1496-1507"},"PeriodicalIF":7.6000,"publicationDate":"2016-02-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5963673/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Chemical Science","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1039/c5sc03447g","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2015/11/24 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
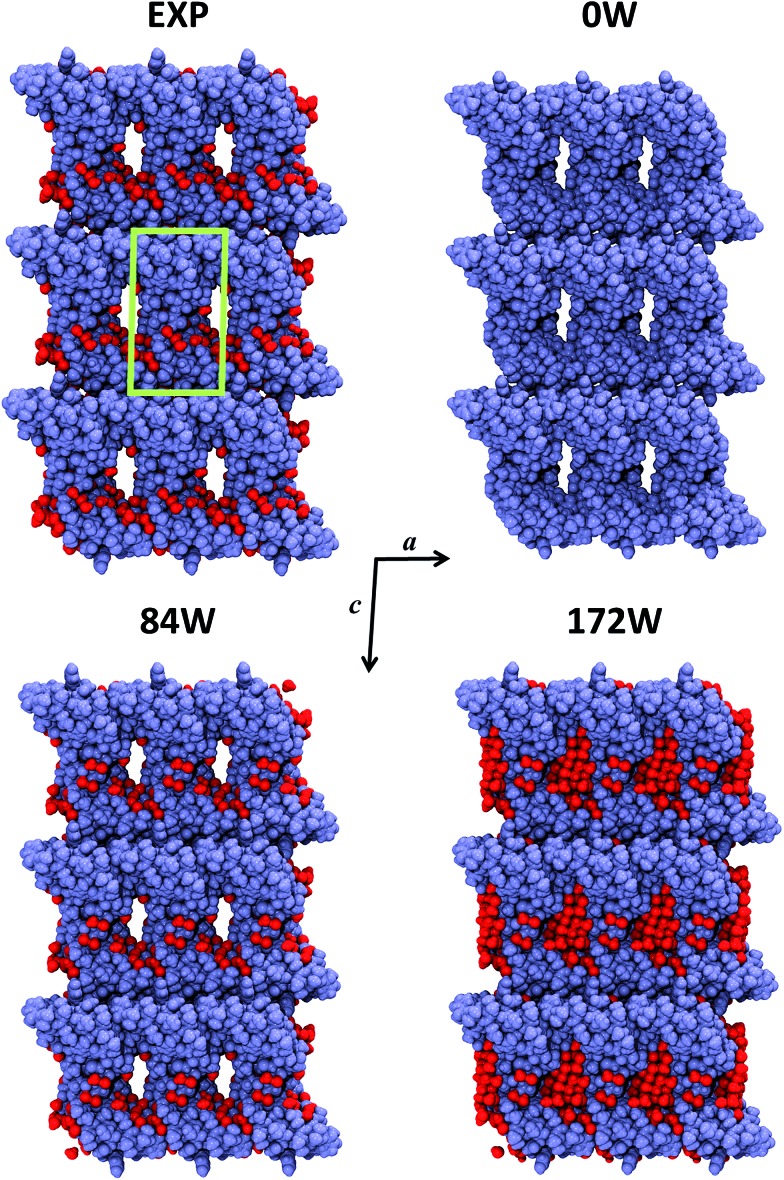
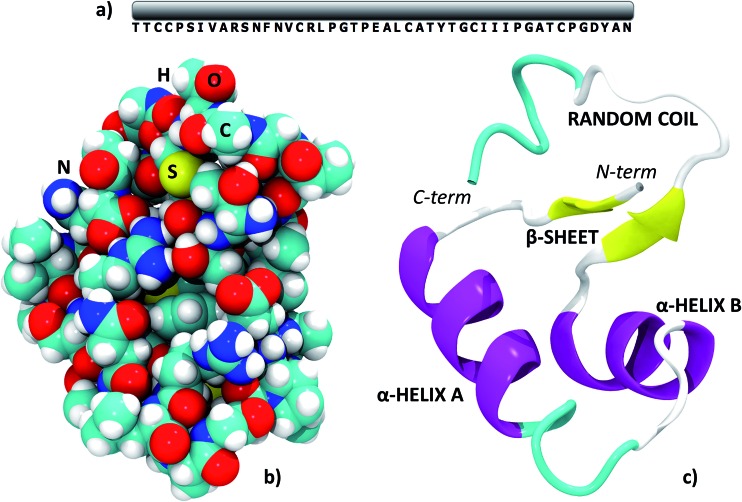
Molecular simulations of proteins have been usually accomplished through empirical or semi-empirical potentials, due to the large size and inherent complexity of these biological systems. On the other hand, a theoretical description of proteins based on quantum-mechanical methods would however provide an unbiased characterization of their electronic properties, possibly offering a link between these and the ultimate biological activity. Yet, such approaches have been historically hindered by the large amount of requested computational power. Here we demonstrate the feasibility of periodic all-electron density functional theory calculations in the description of the crystal of the protein crambin (46 aminoacids), which is determined with exceptional structural accuracy. We have employed the hybrid B3LYP functional, coupled to an empirical description of London interactions (D*) to simulate the crambin crystal with an increasing amount of lattice water molecules in the cell (up to 172H2O per cell). The agreement with the experiment is good for both protein geometry and protein-water interactions. The energetics was computed to predict crystal formation energies, protein-water and protein-protein interaction energies. We studied the role of dispersion interactions which are crucial for holding the crambin crystal in place. B3LYP-D* electrostatic potential and dipole moment of crambin as well as the electronic charge flow from crambin to the solvating water molecules (0.0015e per H2O) have also been predicted. These results proved that quantum-mechanical simulations of small proteins, both free and in their crystalline state, are now feasible in a reasonable amount of time, by programs capable of exploiting high performance computing architectures, allowing the study of protein properties not easily amenable through classical force fields.
期刊介绍:
Chemical Science is a journal that encompasses various disciplines within the chemical sciences. Its scope includes publishing ground-breaking research with significant implications for its respective field, as well as appealing to a wider audience in related areas. To be considered for publication, articles must showcase innovative and original advances in their field of study and be presented in a manner that is understandable to scientists from diverse backgrounds. However, the journal generally does not publish highly specialized research.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


