{"title":"Finding local genome rearrangements.","authors":"Pijus Simonaitis, Krister M Swenson","doi":"10.1186/s13015-018-0127-2","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>The double cut and join (DCJ) model of genome rearrangement is well studied due to its mathematical simplicity and power to account for the many events that transform gene order. These studies have mostly been devoted to the understanding of minimum length scenarios transforming one genome into another. In this paper we search instead for rearrangement scenarios that minimize the number of rearrangements whose breakpoints are unlikely due to some biological criteria. One such criterion has recently become accessible due to the advent of the Hi-C experiment, facilitating the study of 3D spacial distance between breakpoint regions.</p><p><strong>Results: </strong>We establish a link between the minimum number of unlikely rearrangements required by a scenario and the problem of finding a maximum edge-disjoint cycle packing on a certain transformed version of the adjacency graph. This link leads to a 3/2-approximation as well as an exact integer linear programming formulation for our problem, which we prove to be NP-complete. We also present experimental results on fruit flies, showing that Hi-C data is informative when used as a criterion for rearrangements.</p><p><strong>Conclusions: </strong>A new variant of the weighted DCJ distance problem is addressed that ignores scenario length in its objective function. A solution to this problem provides a lower bound on the number of unlikely moves necessary when transforming one gene order into another. This lower bound aids in the study of rearrangement scenarios with respect to chromatin structure, and could eventually be used in the design of a fixed parameter algorithm with a more general objective function.</p>","PeriodicalId":50823,"journal":{"name":"Algorithms for Molecular Biology","volume":"13 ","pages":"9"},"PeriodicalIF":1.5000,"publicationDate":"2018-05-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s13015-018-0127-2","citationCount":"6","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Algorithms for Molecular Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13015-018-0127-2","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2018/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 6
Abstract
Background: The double cut and join (DCJ) model of genome rearrangement is well studied due to its mathematical simplicity and power to account for the many events that transform gene order. These studies have mostly been devoted to the understanding of minimum length scenarios transforming one genome into another. In this paper we search instead for rearrangement scenarios that minimize the number of rearrangements whose breakpoints are unlikely due to some biological criteria. One such criterion has recently become accessible due to the advent of the Hi-C experiment, facilitating the study of 3D spacial distance between breakpoint regions.
Results: We establish a link between the minimum number of unlikely rearrangements required by a scenario and the problem of finding a maximum edge-disjoint cycle packing on a certain transformed version of the adjacency graph. This link leads to a 3/2-approximation as well as an exact integer linear programming formulation for our problem, which we prove to be NP-complete. We also present experimental results on fruit flies, showing that Hi-C data is informative when used as a criterion for rearrangements.
Conclusions: A new variant of the weighted DCJ distance problem is addressed that ignores scenario length in its objective function. A solution to this problem provides a lower bound on the number of unlikely moves necessary when transforming one gene order into another. This lower bound aids in the study of rearrangement scenarios with respect to chromatin structure, and could eventually be used in the design of a fixed parameter algorithm with a more general objective function.
期刊介绍:
Algorithms for Molecular Biology publishes articles on novel algorithms for biological sequence and structure analysis, phylogeny reconstruction, and combinatorial algorithms and machine learning.
Areas of interest include but are not limited to: algorithms for RNA and protein structure analysis, gene prediction and genome analysis, comparative sequence analysis and alignment, phylogeny, gene expression, machine learning, and combinatorial algorithms.
Where appropriate, manuscripts should describe applications to real-world data. However, pure algorithm papers are also welcome if future applications to biological data are to be expected, or if they address complexity or approximation issues of novel computational problems in molecular biology. Articles about novel software tools will be considered for publication if they contain some algorithmically interesting aspects.
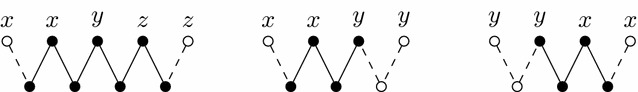
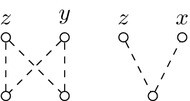
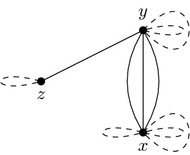

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


