Mubashir Hassan, Qamar Abbas, Hussain Raza, Ahmed A. Moustafa and Sung-Yum Seo
{"title":"Computational analysis of histidine mutations on the structural stability of human tyrosinases leading to albinism insurgence†","authors":"Mubashir Hassan, Qamar Abbas, Hussain Raza, Ahmed A. Moustafa and Sung-Yum Seo","doi":"10.1039/C7MB00211D","DOIUrl":null,"url":null,"abstract":"<p >Misfolding and structural alteration in proteins lead to serious malfunctions and cause various diseases in humans. Mutations at the active binding site in tyrosinase impair structural stability and cause lethal albinism by abolishing copper binding. To evaluate the histidine mutational effect, all mutated structures were built using homology modelling. The protein sequence was retrieved from the UniProt database, and 3D models of original and mutated human tyrosinase sequences were predicted by changing the residual positions within the target sequence separately. Structural and mutational analyses were performed to interpret the significance of mutated residues (N<small><sup>180</sup></small>, R<small><sup>202</sup></small>, Q<small><sup>202</sup></small>, R<small><sup>211</sup></small>, Y<small><sup>363</sup></small>, R<small><sup>367</sup></small>, Y<small><sup>367</sup></small> and D<small><sup>390</sup></small>) at the active binding site of tyrosinases. CSpritz analysis depicted that 23.25% residues actively participate in the instability of tyrosinase. The accuracy of predicted models was confirmed through online servers ProSA-web, ERRAT score and VERIFY 3D values. The theoretical <em>pI</em> and GRAVY generated results also showed the accuracy of the predicted models. The CCA negative correlation results depicted that the replacement of mutated residues at His within the active binding site disturbs the structural stability of tyrosinases. The predicted CCA scores of Tyr<small><sup>367</sup></small> (?0.079) and Q/R<small><sup>202</sup></small> (0.032) revealed that both mutations have more potential to disturb the structural stability. MD simulation analyses of all predicted models justified that Gln<small><sup>202</sup></small>, Arg<small><sup>202</sup></small>, Tyr<small><sup>367</sup></small> and D<small><sup>390</sup></small> replacement made the protein structures more susceptible to destabilization. Mutational results showed that the replacement of His with Q/R<small><sup>202</sup></small> and Y/R<small><sup>363</sup></small> has a lethal effect and may cause melanin associated diseases such as OCA1. Taken together, our computational analysis depicts that the mutated residues such as Q/R<small><sup>202</sup></small> and Y/R<small><sup>363</sup></small> actively participate in instability and misfolding of tyrosinases, which may govern OCA1 through disturbing the melanin biosynthetic pathway.</p>","PeriodicalId":90,"journal":{"name":"Molecular BioSystems","volume":" 8","pages":" 1534-1544"},"PeriodicalIF":3.7430,"publicationDate":"2017-06-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1039/C7MB00211D","citationCount":"12","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular BioSystems","FirstCategoryId":"1085","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2017/mb/c7mb00211d","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 12
Abstract
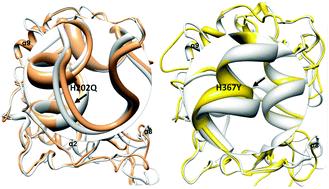
Misfolding and structural alteration in proteins lead to serious malfunctions and cause various diseases in humans. Mutations at the active binding site in tyrosinase impair structural stability and cause lethal albinism by abolishing copper binding. To evaluate the histidine mutational effect, all mutated structures were built using homology modelling. The protein sequence was retrieved from the UniProt database, and 3D models of original and mutated human tyrosinase sequences were predicted by changing the residual positions within the target sequence separately. Structural and mutational analyses were performed to interpret the significance of mutated residues (N180, R202, Q202, R211, Y363, R367, Y367 and D390) at the active binding site of tyrosinases. CSpritz analysis depicted that 23.25% residues actively participate in the instability of tyrosinase. The accuracy of predicted models was confirmed through online servers ProSA-web, ERRAT score and VERIFY 3D values. The theoretical pI and GRAVY generated results also showed the accuracy of the predicted models. The CCA negative correlation results depicted that the replacement of mutated residues at His within the active binding site disturbs the structural stability of tyrosinases. The predicted CCA scores of Tyr367 (?0.079) and Q/R202 (0.032) revealed that both mutations have more potential to disturb the structural stability. MD simulation analyses of all predicted models justified that Gln202, Arg202, Tyr367 and D390 replacement made the protein structures more susceptible to destabilization. Mutational results showed that the replacement of His with Q/R202 and Y/R363 has a lethal effect and may cause melanin associated diseases such as OCA1. Taken together, our computational analysis depicts that the mutated residues such as Q/R202 and Y/R363 actively participate in instability and misfolding of tyrosinases, which may govern OCA1 through disturbing the melanin biosynthetic pathway.
期刊介绍:
Molecular Omics publishes molecular level experimental and bioinformatics research in the -omics sciences, including genomics, proteomics, transcriptomics and metabolomics. We will also welcome multidisciplinary papers presenting studies combining different types of omics, or the interface of omics and other fields such as systems biology or chemical biology.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


