Abdullah H Almalki, Laila F Sadagah, Mohammed Qureshi, Hatim Maghrabi, Abdulrahman Algain, Ahmed Alsaeed
{"title":"Atypical hemolytic-uremic syndrome due to complement factor I mutation.","authors":"Abdullah H Almalki, Laila F Sadagah, Mohammed Qureshi, Hatim Maghrabi, Abdulrahman Algain, Ahmed Alsaeed","doi":"10.5527/wjn.v6.i6.243","DOIUrl":null,"url":null,"abstract":"<p><p>Atypical hemolytic-uremic syndrome (aHUS) is a rare disease of complement dysregulation leading to thrombotic microangiopathy (TMA). Renal involvement and progression to end-stage renal disease are common in untreated patients. We report a 52-year-old female patient who presented with severe acute kidney injury, microangiopathic hemolytic anemia, and thrombocytopenia. She was managed with steroid, plasma exchange, and dialysis. Kidney biopsy shows TMA and renal cortical necrosis. Genetic analysis reveals heterozygous complement factor I (CFI) mutation. Eculizumab was initiated after 3 mo of presentation, continued for 9 mo, and stopped because of sustained hematologic remission, steady renal function, and cost issues. Despite this, the patient continued to be in hematologic remission and showed signs of renal recovery, and peritoneal dialysis was stopped 32 mo after initiation. We report a case of aHUS due to CFI mutation, which, to the best of our knowledge, has not been reported before in Saudi Arabia. Our case illustrates the challenges related to the diagnosis and management of this condition, in which a high index of suspicion and prompt treatment are usually necessary.</p>","PeriodicalId":23745,"journal":{"name":"World Journal of Nephrology","volume":"6 6","pages":"243-250"},"PeriodicalIF":0.0000,"publicationDate":"2017-11-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/8b/3f/WJN-6-243.PMC5714872.pdf","citationCount":"3","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"World Journal of Nephrology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.5527/wjn.v6.i6.243","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 3
Abstract
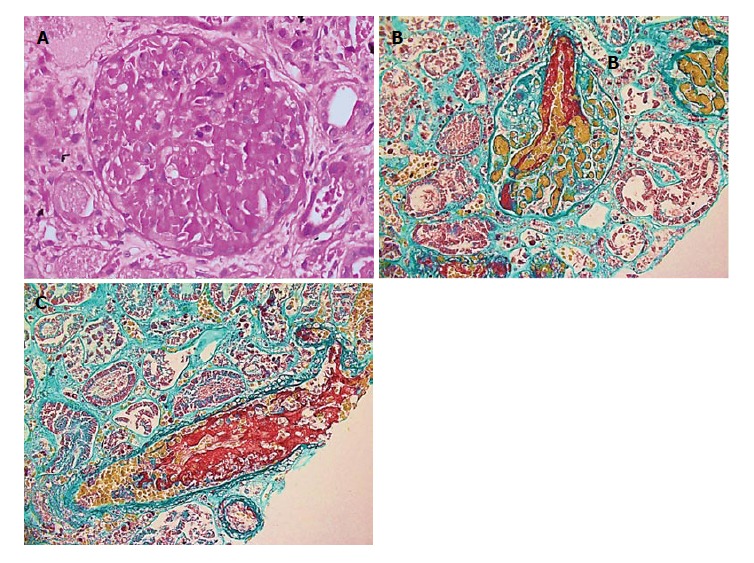
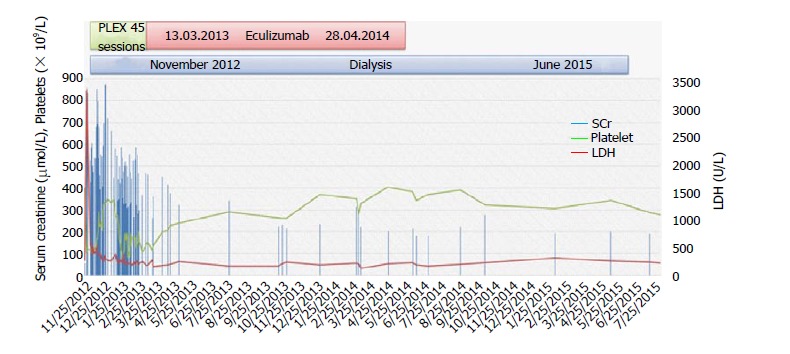
Atypical hemolytic-uremic syndrome (aHUS) is a rare disease of complement dysregulation leading to thrombotic microangiopathy (TMA). Renal involvement and progression to end-stage renal disease are common in untreated patients. We report a 52-year-old female patient who presented with severe acute kidney injury, microangiopathic hemolytic anemia, and thrombocytopenia. She was managed with steroid, plasma exchange, and dialysis. Kidney biopsy shows TMA and renal cortical necrosis. Genetic analysis reveals heterozygous complement factor I (CFI) mutation. Eculizumab was initiated after 3 mo of presentation, continued for 9 mo, and stopped because of sustained hematologic remission, steady renal function, and cost issues. Despite this, the patient continued to be in hematologic remission and showed signs of renal recovery, and peritoneal dialysis was stopped 32 mo after initiation. We report a case of aHUS due to CFI mutation, which, to the best of our knowledge, has not been reported before in Saudi Arabia. Our case illustrates the challenges related to the diagnosis and management of this condition, in which a high index of suspicion and prompt treatment are usually necessary.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


