D S J Groenenberg, J Harl, E Duijm, E Gittenberger
{"title":"The complete mitogenome of <i>Orcula dolium</i> (Draparnaud, 1801); ultra-deep sequencing from a single long-range PCR using the Ion-Torrent PGM.","authors":"D S J Groenenberg, J Harl, E Duijm, E Gittenberger","doi":"10.1186/s41065-017-0028-2","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>With the increasing capacity of present-day next-generation sequencers the field of mitogenomics is rapidly changing. Enrichment of the mitochondrial fraction, is no longer necessary for obtaining mitogenomic data. Despite the benefits, shotgun sequencing approaches also have disadvantages. They do not guarantee obtaining the <i>complete</i> mitogenome, generally require larger amounts of input DNA and coverage is low compared to sequencing with enrichment strategies. If the mitogenome could be amplified in a single amplification, additional time and costs for sample preparation might outweigh these disadvantages.</p><p><strong>Results: </strong>A sequence of the complete mitochondrial genome of the pupilloid landsnail <i>Orcula dolium</i> is presented. The mitogenome was amplified in a single long-range (LR) PCR and sequenced on an Ion Torrent PGM (Life Technologies). The length is 14,063 nt and the average depth of coverage is 1112 X. This is the first published mitogenome for a member of the family Orculidae. It has the typical metazoan makeup of 13 protein coding genes (PCGs), 2 ribosomal RNAs (12S and 16S) and 22 transfer RNAs (tRNAs). <i>Orcula</i> is positioned between <i>Pupilla</i> and the Vertiginidae as the sister-group of <i>Gastrocopta</i> and <i>Vertigo</i>, together. An ancestral gene order reconstruction shows that Orthurethra in contrast to other Stylommatophora, have tRNA-H before tRNA-G and that the gene order in the 'non-achatinoid' clade is identical to that of closely related non-stylommatophoran taxa.</p><p><strong>Conclusions: </strong>We show it is feasible to ultra-deep sequence a mitogenome from a single LR-PCR. This approach is particularly relevant to studies that have low concentrations of input DNA. It results in a more efficient use of NGS capacity (only the targeted fraction is sequenced) and is an effective selection against nuclear mitochondrial inserts (NUMTS). In contrast to previous studies based in particular on 28S, our results indicate that phylogeny reconstructions based on complete mitogenomes might be more suitable to resolve deep relationships within Stylommatophora. Ancestral gene order reconstructions reveal rearrangements that characterize systematic groups.</p>","PeriodicalId":55057,"journal":{"name":"Hereditas","volume":"154 ","pages":"7"},"PeriodicalIF":2.1000,"publicationDate":"2017-04-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5379511/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Hereditas","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s41065-017-0028-2","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2017/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: With the increasing capacity of present-day next-generation sequencers the field of mitogenomics is rapidly changing. Enrichment of the mitochondrial fraction, is no longer necessary for obtaining mitogenomic data. Despite the benefits, shotgun sequencing approaches also have disadvantages. They do not guarantee obtaining the complete mitogenome, generally require larger amounts of input DNA and coverage is low compared to sequencing with enrichment strategies. If the mitogenome could be amplified in a single amplification, additional time and costs for sample preparation might outweigh these disadvantages.
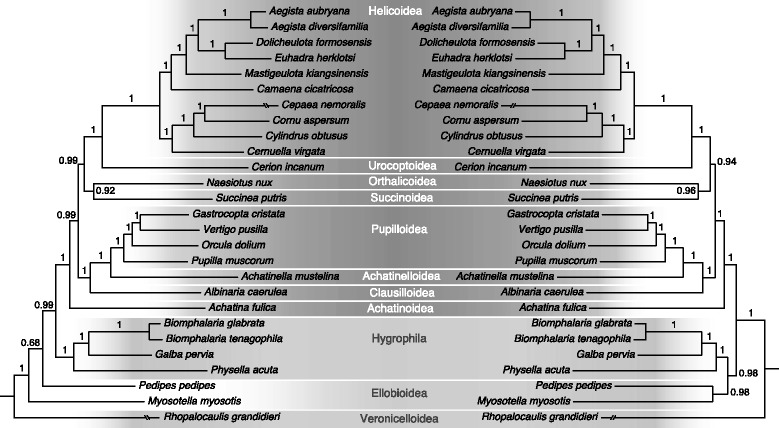
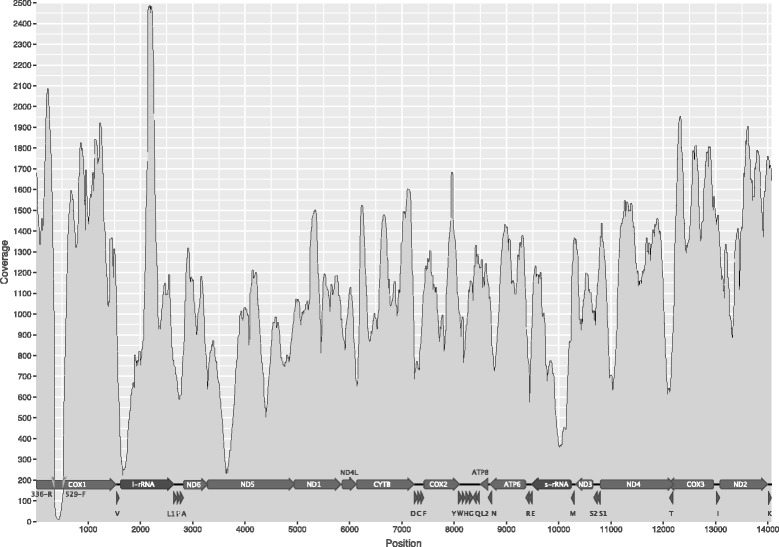
Results: A sequence of the complete mitochondrial genome of the pupilloid landsnail Orcula dolium is presented. The mitogenome was amplified in a single long-range (LR) PCR and sequenced on an Ion Torrent PGM (Life Technologies). The length is 14,063 nt and the average depth of coverage is 1112 X. This is the first published mitogenome for a member of the family Orculidae. It has the typical metazoan makeup of 13 protein coding genes (PCGs), 2 ribosomal RNAs (12S and 16S) and 22 transfer RNAs (tRNAs). Orcula is positioned between Pupilla and the Vertiginidae as the sister-group of Gastrocopta and Vertigo, together. An ancestral gene order reconstruction shows that Orthurethra in contrast to other Stylommatophora, have tRNA-H before tRNA-G and that the gene order in the 'non-achatinoid' clade is identical to that of closely related non-stylommatophoran taxa.
Conclusions: We show it is feasible to ultra-deep sequence a mitogenome from a single LR-PCR. This approach is particularly relevant to studies that have low concentrations of input DNA. It results in a more efficient use of NGS capacity (only the targeted fraction is sequenced) and is an effective selection against nuclear mitochondrial inserts (NUMTS). In contrast to previous studies based in particular on 28S, our results indicate that phylogeny reconstructions based on complete mitogenomes might be more suitable to resolve deep relationships within Stylommatophora. Ancestral gene order reconstructions reveal rearrangements that characterize systematic groups.
期刊介绍:
For almost a century, Hereditas has published original cutting-edge research and reviews. As the Official journal of the Mendelian Society of Lund, the journal welcomes research from across all areas of genetics and genomics. Topics of interest include human and medical genetics, animal and plant genetics, microbial genetics, agriculture and bioinformatics.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


