{"title":"Sustained activation of mTORC1 in macrophages increases AMPKα-dependent autophagy to maintain cellular homeostasis.","authors":"Hongjie Pan, Xiao-Ping Zhong, Sunhee Lee","doi":"10.1186/s12858-016-0069-6","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>The mechanistic target of rapamycin complex 1 (mTORC1) is a well-conserved serine/threonine protein kinase that controls autophagy as well as many other processes such as protein synthesis, cell growth, and metabolism. The activity of mTORC1 is stringently and negatively controlled by the tuberous sclerosis proteins 1 and 2 complex (TSC1/2).</p><p><strong>Results: </strong>In contrast to the previous studies using Tsc1 knockout mouse embryonic fibroblasts (MEF) cells, we demonstrated evidence that TSC1 deficient macrophages exhibited enhanced basal and mycobacterial infection-induced autophagy via AMPKα-dependent phosphorylation of ULK1 (Ser555). These effects were concomitant with constitutive activation of mTORC1 and can be reversed by addition of amino acids or rapamycin, and by the knockdown of the regulatory-associated protein of mTOR, Raptor. In addition, increased autophagy in TSC1 deficient macrophages resulted in suppression of inflammation during mycobacterial infection, which was reversed upon amino acid treatment of the TSC1 deficient macrophages. We further demonstrated that TSC1 conditional knockout mice infected with Mycobacterium tuberculosis, the causative agent of tuberculosis, resulted in less bacterial burden and a comparable level of inflammation when compared to wild type mice.</p><p><strong>Conclusions: </strong>Our data revealed that sustained activation of mTORC1 due to defects in TSC1 promotes AMPKα-dependent autophagic flux to maintain cellular homeostasis.</p>","PeriodicalId":9113,"journal":{"name":"BMC Biochemistry","volume":"17 1","pages":"14"},"PeriodicalIF":0.0000,"publicationDate":"2016-07-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s12858-016-0069-6","citationCount":"21","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Biochemistry","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s12858-016-0069-6","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 21
Abstract
Background: The mechanistic target of rapamycin complex 1 (mTORC1) is a well-conserved serine/threonine protein kinase that controls autophagy as well as many other processes such as protein synthesis, cell growth, and metabolism. The activity of mTORC1 is stringently and negatively controlled by the tuberous sclerosis proteins 1 and 2 complex (TSC1/2).
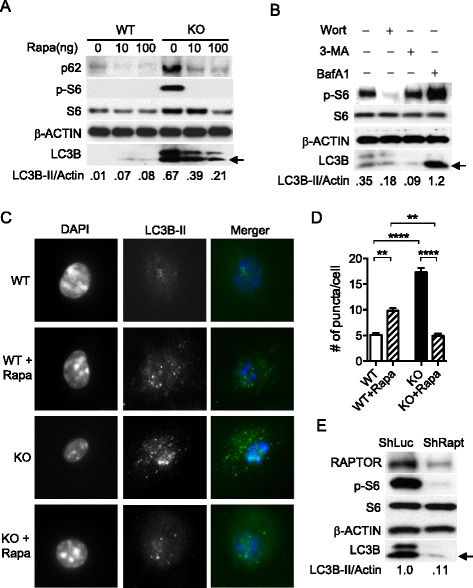
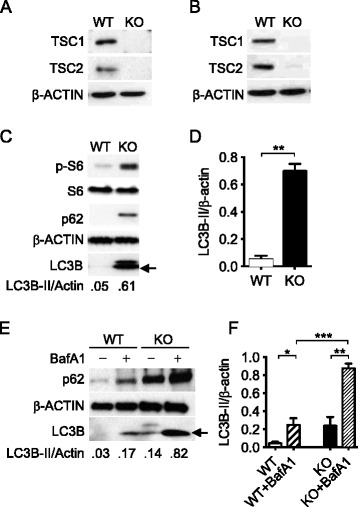
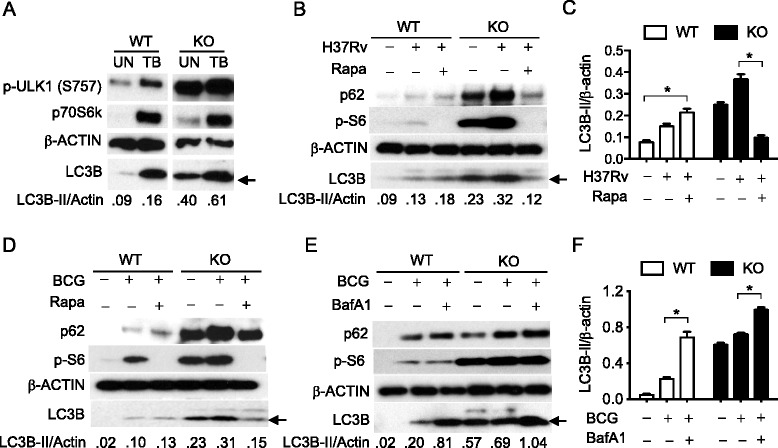
Results: In contrast to the previous studies using Tsc1 knockout mouse embryonic fibroblasts (MEF) cells, we demonstrated evidence that TSC1 deficient macrophages exhibited enhanced basal and mycobacterial infection-induced autophagy via AMPKα-dependent phosphorylation of ULK1 (Ser555). These effects were concomitant with constitutive activation of mTORC1 and can be reversed by addition of amino acids or rapamycin, and by the knockdown of the regulatory-associated protein of mTOR, Raptor. In addition, increased autophagy in TSC1 deficient macrophages resulted in suppression of inflammation during mycobacterial infection, which was reversed upon amino acid treatment of the TSC1 deficient macrophages. We further demonstrated that TSC1 conditional knockout mice infected with Mycobacterium tuberculosis, the causative agent of tuberculosis, resulted in less bacterial burden and a comparable level of inflammation when compared to wild type mice.
Conclusions: Our data revealed that sustained activation of mTORC1 due to defects in TSC1 promotes AMPKα-dependent autophagic flux to maintain cellular homeostasis.
期刊介绍:
BMC Biochemistry is an open access journal publishing original peer-reviewed research articles in all aspects of biochemical processes, including the structure, function and dynamics of metabolic pathways, supramolecular complexes, enzymes, proteins, nucleic acids and small molecular components of organelles, cells and tissues. BMC Biochemistry (ISSN 1471-2091) is indexed/tracked/covered by PubMed, MEDLINE, BIOSIS, CAS, EMBASE, Scopus, Zoological Record, Thomson Reuters (ISI) and Google Scholar.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


