{"title":"Identifying Potential Regions of Copy Number Variation for Bipolar Disorder.","authors":"Yi-Hsuan Chen, Ru-Band Lu, Hung Hung, Po-Hsiu Kuo","doi":"10.3390/microarrays3010052","DOIUrl":null,"url":null,"abstract":"<p><p>Bipolar disorder is a complex psychiatric disorder with high heritability, but its genetic determinants are still largely unknown. Copy number variation (CNV) is one of the sources to explain part of the heritability. However, it is a challenge to estimate discrete values of the copy numbers using continuous signals calling from a set of markers, and to simultaneously perform association testing between CNVs and phenotypic outcomes. The goal of the present study is to perform a series of data filtering and analysis procedures using a DNA pooling strategy to identify potential CNV regions that are related to bipolar disorder. A total of 200 normal controls and 200 clinically diagnosed bipolar patients were recruited in this study, and were randomly divided into eight control and eight case pools. Genome-wide genotyping was employed using Illumina Human Omni1-Quad array with approximately one million markers for CNV calling. We aimed at setting a series of criteria to filter out the signal noise of marker data and to reduce the chance of false-positive findings for CNV regions. We first defined CNV regions for each pool. Potential CNV regions were reported based on the different patterns of CNV status between cases and controls. Genes that were mapped into the potential CNV regions were examined with association testing, Gene Ontology enrichment analysis, and checked with existing literature for their associations with bipolar disorder. We reported several CNV regions that are related to bipolar disorder. Two CNV regions on chromosome 11 and 22 showed significant signal differences between cases and controls (p < 0.05). Another five CNV regions on chromosome 6, 9, and 19 were overlapped with results in previous CNV studies. Experimental validation of two CNV regions lent some support to our reported findings. Further experimental and replication studies could be designed for these selected regions. </p>","PeriodicalId":56355,"journal":{"name":"Microarrays","volume":"3 1","pages":"52-71"},"PeriodicalIF":0.0000,"publicationDate":"2014-02-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.3390/microarrays3010052","citationCount":"6","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Microarrays","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3390/microarrays3010052","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 6
Abstract
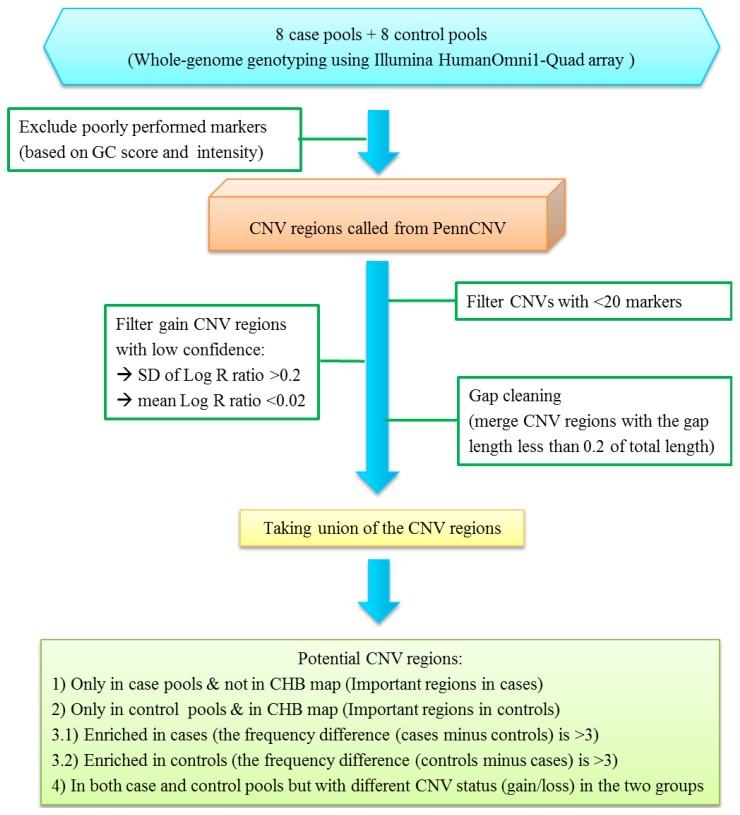
Bipolar disorder is a complex psychiatric disorder with high heritability, but its genetic determinants are still largely unknown. Copy number variation (CNV) is one of the sources to explain part of the heritability. However, it is a challenge to estimate discrete values of the copy numbers using continuous signals calling from a set of markers, and to simultaneously perform association testing between CNVs and phenotypic outcomes. The goal of the present study is to perform a series of data filtering and analysis procedures using a DNA pooling strategy to identify potential CNV regions that are related to bipolar disorder. A total of 200 normal controls and 200 clinically diagnosed bipolar patients were recruited in this study, and were randomly divided into eight control and eight case pools. Genome-wide genotyping was employed using Illumina Human Omni1-Quad array with approximately one million markers for CNV calling. We aimed at setting a series of criteria to filter out the signal noise of marker data and to reduce the chance of false-positive findings for CNV regions. We first defined CNV regions for each pool. Potential CNV regions were reported based on the different patterns of CNV status between cases and controls. Genes that were mapped into the potential CNV regions were examined with association testing, Gene Ontology enrichment analysis, and checked with existing literature for their associations with bipolar disorder. We reported several CNV regions that are related to bipolar disorder. Two CNV regions on chromosome 11 and 22 showed significant signal differences between cases and controls (p < 0.05). Another five CNV regions on chromosome 6, 9, and 19 were overlapped with results in previous CNV studies. Experimental validation of two CNV regions lent some support to our reported findings. Further experimental and replication studies could be designed for these selected regions.
双相情感障碍是一种具有高遗传性的复杂精神障碍,但其遗传决定因素在很大程度上仍是未知的。拷贝数变异(CNV)是解释部分遗传力的来源之一。然而,使用来自一组标记的连续信号来估计拷贝数的离散值,并同时执行CNVs与表型结果之间的关联测试是一个挑战。本研究的目的是使用DNA池策略执行一系列数据过滤和分析程序,以确定与双相情感障碍相关的潜在CNV区域。本研究共招募200名正常对照和200名临床诊断的双相情感障碍患者,随机分为8个对照和8个病例池。使用Illumina Human Omni1-Quad阵列进行全基因组分型,该阵列具有大约100万个CNV调用标记。我们的目标是建立一系列标准来过滤掉标记数据的信号噪声,并减少CNV区域假阳性结果的机会。我们首先为每个池定义CNV区域。根据病例和对照组之间CNV状态的不同模式,报告了潜在的CNV区域。绘制到潜在CNV区域的基因通过关联测试、基因本体富集分析进行检查,并与现有文献核对其与双相情感障碍的关联。我们报道了几个与双相情感障碍相关的CNV区域。11号和22号染色体上的两个CNV区与对照组差异有统计学意义(p < 0.05)。6号、9号和19号染色体上的另外5个CNV区域与之前的CNV研究结果重叠。两个CNV区域的实验验证为我们报道的发现提供了一些支持。可以为这些选定的区域设计进一步的实验和复制研究。
期刊介绍:
High-Throughput (formerly Microarrays, ISSN 2076-3905) is a multidisciplinary peer-reviewed scientific journal that provides an advanced forum for the publication of studies reporting high-dimensional approaches and developments in Life Sciences, Chemistry and related fields. Our aim is to encourage scientists to publish their experimental and theoretical results based on high-throughput techniques as well as computational and statistical tools for data analysis and interpretation. The full experimental or methodological details must be provided so that the results can be reproduced. There is no restriction on the length of the papers. High-Throughput invites submissions covering several topics, including, but not limited to: Microarrays, DNA Sequencing, RNA Sequencing, Protein Identification and Quantification, Cell-based Approaches, Omics Technologies, Imaging, Bioinformatics, Computational Biology/Chemistry, Statistics, Integrative Omics, Drug Discovery and Development, Microfluidics, Lab-on-a-chip, Data Mining, Databases, Multiplex Assays.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


