Jean-Loup Duband, Sophie Escot, Claire Fournier-Thibault
{"title":"SDF1-CXCR4 signaling: A new player involved in DiGeorge/22q11-deletion syndrome.","authors":"Jean-Loup Duband, Sophie Escot, Claire Fournier-Thibault","doi":"10.1080/21675511.2016.1195050","DOIUrl":null,"url":null,"abstract":"<p><p>The DiGeorge/22q11-deletion syndrome (22q11DS), also known as velocardiofacial syndrome, is a congenital disease causing numerous structural and behavioral disorders, including cardiac outflow tract anomalies, craniofacial dysmorphogenesis, parathyroid and thymus hypoplasia, and mental disorders. It results from a unique chromosomal microdeletion on the 22q11.2 region in which the transcriptional activator TBX1 is decisive for the occurrence of the disease. During embryogenesis, Tbx1 is required for patterning of pharyngeal region giving rise to structures of the face, neck and chest. Genetic and developmental studies demonstrated that the severity and variability of the syndrome are determined by Tbx1 targets involved in pharyngeal neural crest cell migration and survival. Recently, we demonstrated that the chemokine Sdf1/Cxcl12 and its receptor Cxcr4 are genetically downstream of Tbx1 during pharyngeal development and that reduction of CXCR4 signaling results in defects which recapitulate the major morphological anomalies of 22q11DS, supporting the possibility of a pivotal role for the SDF1/CXCR4 axis in its etiology. </p>","PeriodicalId":74639,"journal":{"name":"Rare diseases (Austin, Tex.)","volume":"4 1","pages":"e1195050"},"PeriodicalIF":0.0000,"publicationDate":"2016-06-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1080/21675511.2016.1195050","citationCount":"9","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Rare diseases (Austin, Tex.)","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1080/21675511.2016.1195050","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2016/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 9
Abstract
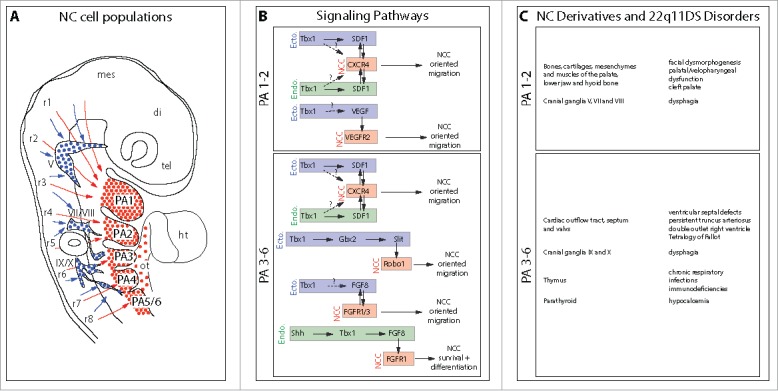
The DiGeorge/22q11-deletion syndrome (22q11DS), also known as velocardiofacial syndrome, is a congenital disease causing numerous structural and behavioral disorders, including cardiac outflow tract anomalies, craniofacial dysmorphogenesis, parathyroid and thymus hypoplasia, and mental disorders. It results from a unique chromosomal microdeletion on the 22q11.2 region in which the transcriptional activator TBX1 is decisive for the occurrence of the disease. During embryogenesis, Tbx1 is required for patterning of pharyngeal region giving rise to structures of the face, neck and chest. Genetic and developmental studies demonstrated that the severity and variability of the syndrome are determined by Tbx1 targets involved in pharyngeal neural crest cell migration and survival. Recently, we demonstrated that the chemokine Sdf1/Cxcl12 and its receptor Cxcr4 are genetically downstream of Tbx1 during pharyngeal development and that reduction of CXCR4 signaling results in defects which recapitulate the major morphological anomalies of 22q11DS, supporting the possibility of a pivotal role for the SDF1/CXCR4 axis in its etiology.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


