Crosstalk between 2 organelles: Lysosomal storage of heparan sulfate causes mitochondrial defects and neuronal death in mucopolysaccharidosis III type C.
{"title":"Crosstalk between 2 organelles: Lysosomal storage of heparan sulfate causes mitochondrial defects and neuronal death in mucopolysaccharidosis III type C.","authors":"Alexey V Pshezhetsky","doi":"10.1080/21675511.2015.1049793","DOIUrl":null,"url":null,"abstract":"<p><p>More than 30% of all lysosomal diseases are mucopolysaccharidoses, disorders affecting the enzymes needed for the stepwise degradation of glycosaminoglycans (mucopolysaccharides). Mucopolysaccharidosis type IIIC (MPS IIIC) is a severe neurologic disease caused by genetic deficiency of heparan sulfate acetyl-CoA: α-glucosaminide N-acetyltransferase (HGSNAT). Through our studies, we have cloned the gene, identified molecular defects in MPS IIIC patients and most recently completed phenotypic characterization of the first animal model of the disease, a mouse with a germline inactivation of the Hgsnat gene.(1) The obtained data have led us to propose that Hgsnat deficiency and lysosomal accumulation of heparan sulfate in microglial cells followed by their activation and cytokine release result in mitochondrial dysfunction in the neurons causing their death which explains why MPS IIIC manifests primarily as a neurodegenerative disease. The goal of this addendum is to summarize data yielding new insights into the mechanism of MPS IIIC and promising novel therapeutic solutions for this and similar disorders. </p>","PeriodicalId":74639,"journal":{"name":"Rare diseases (Austin, Tex.)","volume":"3 1","pages":"e1049793"},"PeriodicalIF":0.0000,"publicationDate":"2015-05-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1080/21675511.2015.1049793","citationCount":"25","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Rare diseases (Austin, Tex.)","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1080/21675511.2015.1049793","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2015/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 25
Abstract
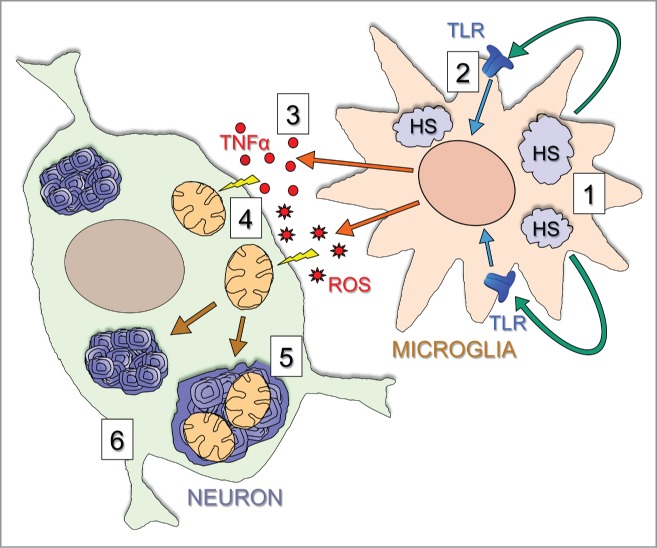
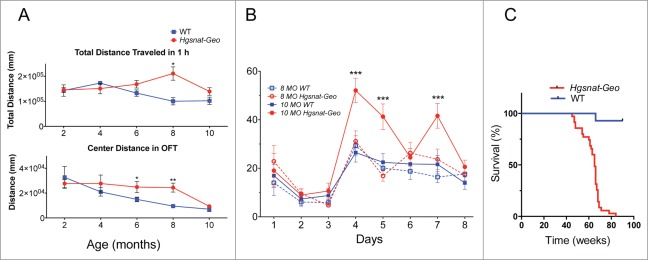
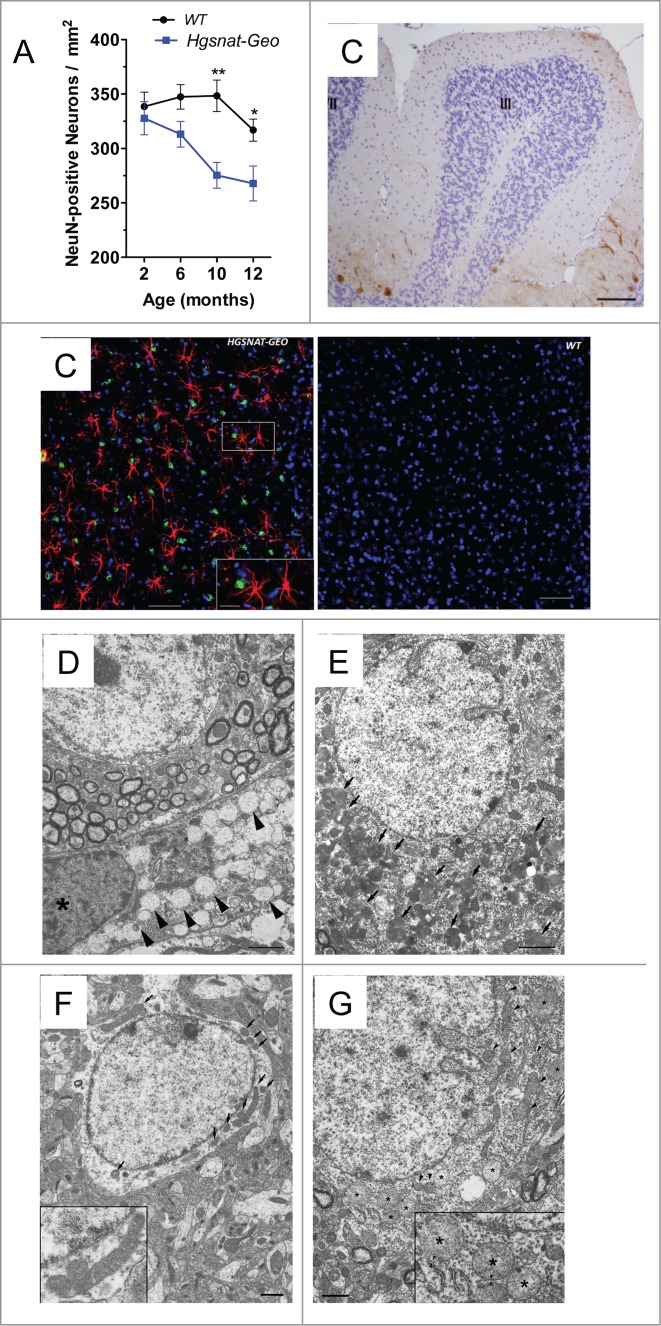
More than 30% of all lysosomal diseases are mucopolysaccharidoses, disorders affecting the enzymes needed for the stepwise degradation of glycosaminoglycans (mucopolysaccharides). Mucopolysaccharidosis type IIIC (MPS IIIC) is a severe neurologic disease caused by genetic deficiency of heparan sulfate acetyl-CoA: α-glucosaminide N-acetyltransferase (HGSNAT). Through our studies, we have cloned the gene, identified molecular defects in MPS IIIC patients and most recently completed phenotypic characterization of the first animal model of the disease, a mouse with a germline inactivation of the Hgsnat gene.(1) The obtained data have led us to propose that Hgsnat deficiency and lysosomal accumulation of heparan sulfate in microglial cells followed by their activation and cytokine release result in mitochondrial dysfunction in the neurons causing their death which explains why MPS IIIC manifests primarily as a neurodegenerative disease. The goal of this addendum is to summarize data yielding new insights into the mechanism of MPS IIIC and promising novel therapeutic solutions for this and similar disorders.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


