Pathogenic Novel Heterozygous Variant c.1076c>T p. (Ser359Phe) chr1: 120512166 in NOTCH2 Gene, Type 2 Alagille Syndrome Causing Neonatal Cholestasis: A Case Report.
Mohammed Shahab Uddin, Saleh Al Fulayyih, Fatin Fahad Al Denaini, Maher Mohammed Al Hatlani
{"title":"Pathogenic Novel Heterozygous Variant c.1076c>T p. (Ser359Phe) chr1: 120512166 in NOTCH2 Gene, Type 2 Alagille Syndrome Causing Neonatal Cholestasis: A Case Report.","authors":"Mohammed Shahab Uddin, Saleh Al Fulayyih, Fatin Fahad Al Denaini, Maher Mohammed Al Hatlani","doi":"10.12659/AJCR.935840","DOIUrl":null,"url":null,"abstract":"<p><p>BACKGROUND Alagille syndrome (ALGS) is a multisystem hereditary illness with a dominant pattern and partial penetrance. Multiple organ abnormalities can be caused by mutations in the Jagged canonical Notch ligand 1 (JAG1) gene. Notch receptor 2 (NOTCH2) gene mutations are also uncommon. ALGS is also characterized by deformed or narrowed bile ducts and is notoriously difficult to diagnose due to the wide range of symptoms and absence of unambiguous genotype-phenotype connections. Little is known about ALGS patients who have NOTCH2 mutations. We present a patient who developed progressive liver failure due to a unique pathogenic heterozygous variation of the NOTCH2 gene, c.1076c>T p. (Ser359Phe) chr1: 120512166, resulting in type 2 ALGS. CASE REPORT A Saudi Arabian newborn with bilateral hazy eyes, ectropion, dry ichthyic skin, normal male genitalia, and bilateral undescended testes was born at 31 weeks. Previous miscarriages, pregnancy-induced maternal cholestasis, fatty liver, or neonatal jaundice were not reported in the family history. He had developed worsening cholestatic jaundice by the third week of hospitalization. The extensive work-up for metabolic, infectious, and other relevant etiologies was negative. Following gram-negative sepsis, he died of multiorgan failure. A NOTCH2 gene mutation explained the phenotypic difference in our situation. Another intriguing observation was the presence of ichthysis and craniosynostosis in ALGS with a NOTCH2 mutation. CONCLUSIONS Cholestasis in newborns can be difficult to diagnose. Next-generation sequencing detects 112 copy number variants in the cholestasis gene panel blood test. More research is needed to understand why NOTCH2 mutations are relatively rare in ALGS.</p>","PeriodicalId":205256,"journal":{"name":"The American Journal of Case Reports","volume":" ","pages":"e935840"},"PeriodicalIF":0.0000,"publicationDate":"2022-10-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/8e/77/amjcaserep-23-e935840.PMC9552858.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The American Journal of Case Reports","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.12659/AJCR.935840","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
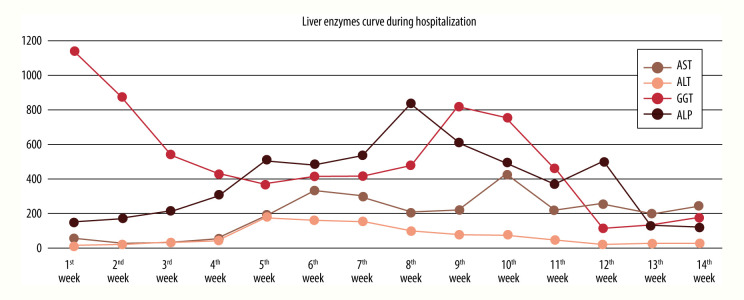
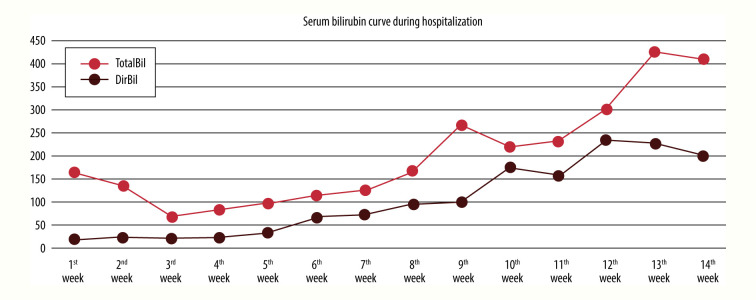
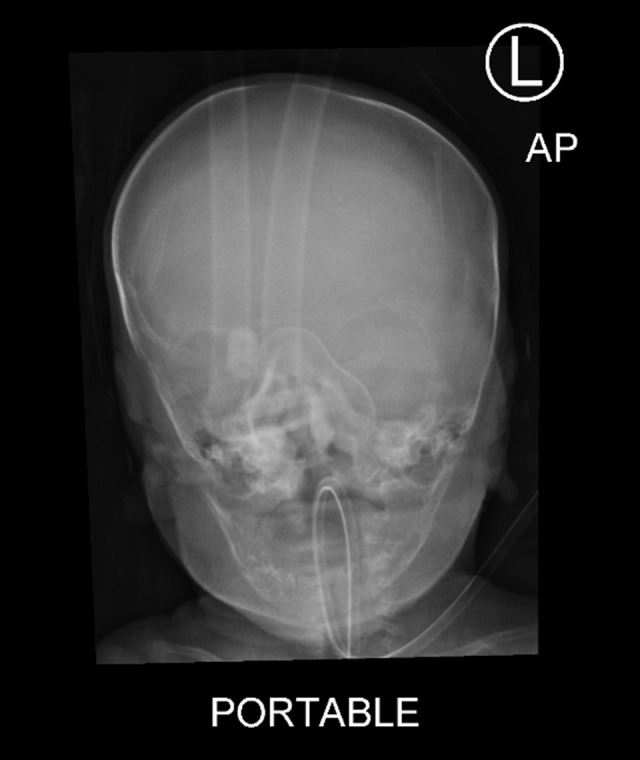
BACKGROUND Alagille syndrome (ALGS) is a multisystem hereditary illness with a dominant pattern and partial penetrance. Multiple organ abnormalities can be caused by mutations in the Jagged canonical Notch ligand 1 (JAG1) gene. Notch receptor 2 (NOTCH2) gene mutations are also uncommon. ALGS is also characterized by deformed or narrowed bile ducts and is notoriously difficult to diagnose due to the wide range of symptoms and absence of unambiguous genotype-phenotype connections. Little is known about ALGS patients who have NOTCH2 mutations. We present a patient who developed progressive liver failure due to a unique pathogenic heterozygous variation of the NOTCH2 gene, c.1076c>T p. (Ser359Phe) chr1: 120512166, resulting in type 2 ALGS. CASE REPORT A Saudi Arabian newborn with bilateral hazy eyes, ectropion, dry ichthyic skin, normal male genitalia, and bilateral undescended testes was born at 31 weeks. Previous miscarriages, pregnancy-induced maternal cholestasis, fatty liver, or neonatal jaundice were not reported in the family history. He had developed worsening cholestatic jaundice by the third week of hospitalization. The extensive work-up for metabolic, infectious, and other relevant etiologies was negative. Following gram-negative sepsis, he died of multiorgan failure. A NOTCH2 gene mutation explained the phenotypic difference in our situation. Another intriguing observation was the presence of ichthysis and craniosynostosis in ALGS with a NOTCH2 mutation. CONCLUSIONS Cholestasis in newborns can be difficult to diagnose. Next-generation sequencing detects 112 copy number variants in the cholestasis gene panel blood test. More research is needed to understand why NOTCH2 mutations are relatively rare in ALGS.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


