Hikmet F Nural-Guvener, Luidmila Zakharova, James Nimlos, Snjezana Popovic, Diego Mastroeni, Mohamed A Gaballa
{"title":"HDAC class I inhibitor, Mocetinostat, reverses cardiac fibrosis in heart failure and diminishes CD90+ cardiac myofibroblast activation.","authors":"Hikmet F Nural-Guvener, Luidmila Zakharova, James Nimlos, Snjezana Popovic, Diego Mastroeni, Mohamed A Gaballa","doi":"10.1186/1755-1536-7-10","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Interstitial fibrosis and fibrotic scar formation contribute to cardiac remodeling and loss of cardiac function in myocardial infarction (MI) and heart failure. Recent studies showed that histone deacetylase (HDAC) inhibitors retard fibrosis formation in acute MI settings. However, it is unknown whether HDAC inhibition can reverse cardiac fibrosis in ischemic heart failure. In addition, specific HDAC isoforms involved in cardiac fibrosis and myofibroblast activation are not well defined. Thus, the purpose of this study is to determine the effects of selective class I HDAC inhibition on cardiac fibroblasts activation and cardiac fibrosis in a congestive heart failure (CHF) model secondary to MI.</p><p><strong>Methods: </strong>MI was created by left anterior descending (LAD) coronary artery occlusion. Class I HDACs were selectively inhibited via Mocetinostat in CD90+ fibroblasts isolated from atrial and ventricular heart tissue in vitro. In vivo, Class I HDACs were inhibited in 3 weeks post MI rats by injecting Mocetinostat for the duration of 3 weeks. Cardiac function and heart tissue were analyzed at 6 weeks post MI.</p><p><strong>Results: </strong>In sham hearts, HDAC1 and HDAC2 displayed differential expression patterns where HDAC1 mainly expressed in cardiac fibroblast and HDAC2 in cardiomyocytes. On the other hand, we showed that HDAC1 and 2 were upregulated in CHF hearts, and were found to co-localize with CD90+ cardiac fibroblasts. In vivo treatment of CHF animals with Mocetinostat improved left ventricle end diastolic pressure and dp/dt max and decreased the total collagen amount. In vitro treatment of CD90+ cells with Mocetinostat reversed myofibroblast phenotype as indicated by a decrease in α-Smooth muscle actin (α-SMA), Collagen III, and Matrix metalloproteinase-2 (MMP2). Furthermore, Mocetinostat increased E-cadherin, induced β-catenin localization to the membrane, and reduced Akt/GSK3β signaling in atrial cardiac fibroblasts. In addition, Mocetinostat treatment of atrial CD90+ cells upregulated cleaved-Caspase3 and activated the p53/p21 axis.</p><p><strong>Conclusions: </strong>Taken together, our results demonstrate upregulation of HDAC1 and 2 in CHF. In addition, HDAC inhibition reverses interstitial fibrosis in CHF. Possible anti-fibrotic actions of HDAC inhibition include reversal of myofibroblast activation and induction of cell cycle arrest/apoptosis.</p>","PeriodicalId":12264,"journal":{"name":"Fibrogenesis & Tissue Repair","volume":"7 ","pages":"10"},"PeriodicalIF":0.0000,"publicationDate":"2014-07-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4094898/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Fibrogenesis & Tissue Repair","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/1755-1536-7-10","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2014/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Interstitial fibrosis and fibrotic scar formation contribute to cardiac remodeling and loss of cardiac function in myocardial infarction (MI) and heart failure. Recent studies showed that histone deacetylase (HDAC) inhibitors retard fibrosis formation in acute MI settings. However, it is unknown whether HDAC inhibition can reverse cardiac fibrosis in ischemic heart failure. In addition, specific HDAC isoforms involved in cardiac fibrosis and myofibroblast activation are not well defined. Thus, the purpose of this study is to determine the effects of selective class I HDAC inhibition on cardiac fibroblasts activation and cardiac fibrosis in a congestive heart failure (CHF) model secondary to MI.
Methods: MI was created by left anterior descending (LAD) coronary artery occlusion. Class I HDACs were selectively inhibited via Mocetinostat in CD90+ fibroblasts isolated from atrial and ventricular heart tissue in vitro. In vivo, Class I HDACs were inhibited in 3 weeks post MI rats by injecting Mocetinostat for the duration of 3 weeks. Cardiac function and heart tissue were analyzed at 6 weeks post MI.
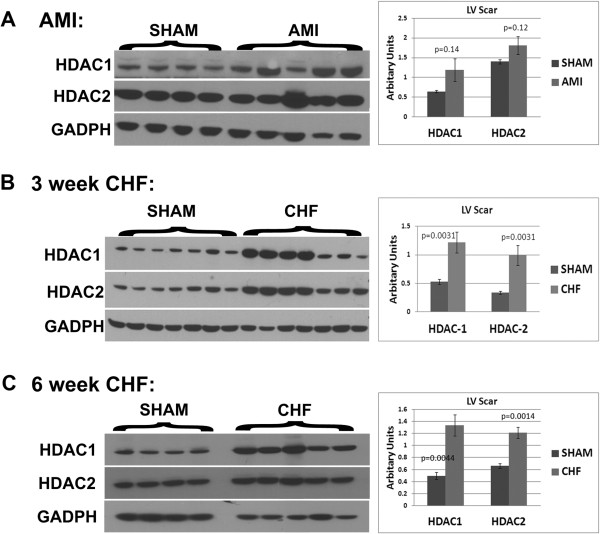
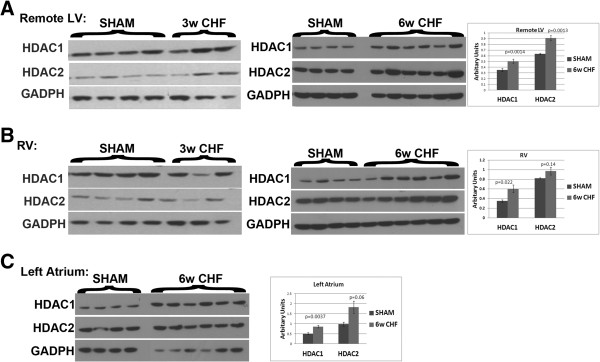
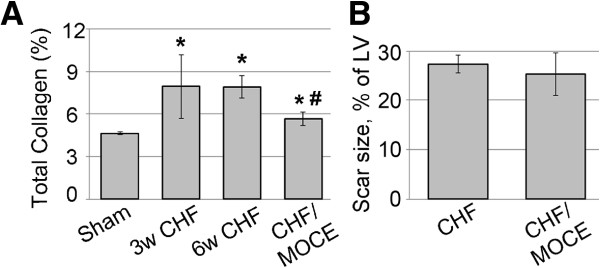
Results: In sham hearts, HDAC1 and HDAC2 displayed differential expression patterns where HDAC1 mainly expressed in cardiac fibroblast and HDAC2 in cardiomyocytes. On the other hand, we showed that HDAC1 and 2 were upregulated in CHF hearts, and were found to co-localize with CD90+ cardiac fibroblasts. In vivo treatment of CHF animals with Mocetinostat improved left ventricle end diastolic pressure and dp/dt max and decreased the total collagen amount. In vitro treatment of CD90+ cells with Mocetinostat reversed myofibroblast phenotype as indicated by a decrease in α-Smooth muscle actin (α-SMA), Collagen III, and Matrix metalloproteinase-2 (MMP2). Furthermore, Mocetinostat increased E-cadherin, induced β-catenin localization to the membrane, and reduced Akt/GSK3β signaling in atrial cardiac fibroblasts. In addition, Mocetinostat treatment of atrial CD90+ cells upregulated cleaved-Caspase3 and activated the p53/p21 axis.
Conclusions: Taken together, our results demonstrate upregulation of HDAC1 and 2 in CHF. In addition, HDAC inhibition reverses interstitial fibrosis in CHF. Possible anti-fibrotic actions of HDAC inhibition include reversal of myofibroblast activation and induction of cell cycle arrest/apoptosis.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


