Lea Tuzovic, Lan Yu, Wenqi Zeng, Xiang Li, Hong Lu, Hsiao-Mei Lu, Kelly Df Gonzalez, Wendy K Chung
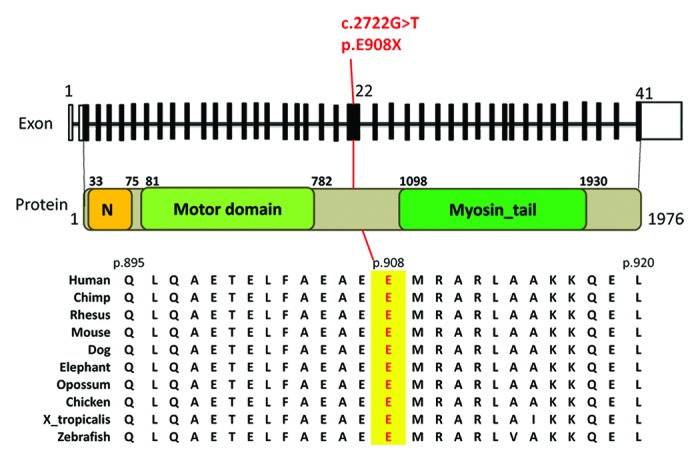
{"title":"A human de novo mutation in MYH10 phenocopies the loss of function mutation in mice.","authors":"Lea Tuzovic, Lan Yu, Wenqi Zeng, Xiang Li, Hong Lu, Hsiao-Mei Lu, Kelly Df Gonzalez, Wendy K Chung","doi":"10.4161/rdis.26144","DOIUrl":null,"url":null,"abstract":"<p><p>We used whole exome sequence analysis to investigate a possible genetic etiology for a patient with the phenotype of intrauterine growth restriction, microcephaly, developmental delay, failure to thrive, congenital bilateral hip dysplasia, cerebral and cerebellar atrophy, hydrocephalus, and congenital diaphragmatic hernia (CDH). Whole exome sequencing identified a novel de novo c.2722G > T (p.E908X) mutation in the Myosin Heavy Chain 10 gene (MYH10) which encodes for non-muscle heavy chain II B (NMHC IIB). Mutations in MYH10 have not been previously described in association with human disease. The E908X mutation is located in the coiled-coil region of the protein and is expected to delete the tail domain and disrupt filament assembly. Nonmuscle myosin IIs (NM IIs) are a group of ubiquitously expressed proteins, and NM II B is specifically enriched in neuronal tissue and is thought to be important in neuronal migration. It is also expressed in cardiac myocytes along with NM IIC. Homozygous NMHC II B-/B- mouse knockouts die by embryonic day (E)14.5 with severe cardiac defects (membranous ventricular septal defect and cardiac outflow tract abnormalities) and neurodevelopmental disorders (progressive hydrocephalus and neuronal migrational abnormalities). A heterozygous MYH10 loss of function mutation produces a severe neurologic phenotype and CDH but no apparent cardiac phenotype and suggests that MYH10 may represent a novel gene for brain malformations and/or CDH. </p>","PeriodicalId":74639,"journal":{"name":"Rare diseases (Austin, Tex.)","volume":"1 ","pages":"e26144"},"PeriodicalIF":0.0000,"publicationDate":"2013-08-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.4161/rdis.26144","citationCount":"40","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Rare diseases (Austin, Tex.)","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.4161/rdis.26144","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2013/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 40
Abstract
We used whole exome sequence analysis to investigate a possible genetic etiology for a patient with the phenotype of intrauterine growth restriction, microcephaly, developmental delay, failure to thrive, congenital bilateral hip dysplasia, cerebral and cerebellar atrophy, hydrocephalus, and congenital diaphragmatic hernia (CDH). Whole exome sequencing identified a novel de novo c.2722G > T (p.E908X) mutation in the Myosin Heavy Chain 10 gene (MYH10) which encodes for non-muscle heavy chain II B (NMHC IIB). Mutations in MYH10 have not been previously described in association with human disease. The E908X mutation is located in the coiled-coil region of the protein and is expected to delete the tail domain and disrupt filament assembly. Nonmuscle myosin IIs (NM IIs) are a group of ubiquitously expressed proteins, and NM II B is specifically enriched in neuronal tissue and is thought to be important in neuronal migration. It is also expressed in cardiac myocytes along with NM IIC. Homozygous NMHC II B-/B- mouse knockouts die by embryonic day (E)14.5 with severe cardiac defects (membranous ventricular septal defect and cardiac outflow tract abnormalities) and neurodevelopmental disorders (progressive hydrocephalus and neuronal migrational abnormalities). A heterozygous MYH10 loss of function mutation produces a severe neurologic phenotype and CDH but no apparent cardiac phenotype and suggests that MYH10 may represent a novel gene for brain malformations and/or CDH.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


