{"title":"Explaining intermediate filament accumulation in giant axonal neuropathy.","authors":"Puneet Opal, Robert D Goldman","doi":"10.4161/rdis.25378","DOIUrl":null,"url":null,"abstract":"<p><p>Giant axonal neuropathy (GAN)(1) is a rare autosomal recessive neurological disorder caused by mutations in the GAN gene that encodes gigaxonin, a member of the BTB/Kelch family of E3 ligase adaptor proteins.(1) This disease is characterized by the aggregation of Intermediate Filaments (IF)-cytoskeletal elements that play important roles in cell physiology including the regulation of cell shape, motility, mechanics and intra-cellular signaling. Although a range of cell types are affected in GAN, neurons display the most severe pathology, with neuronal intermediate filament accumulation and aggregation; this in turn causes axonal swellings or \"giant axons.\" A mechanistic understanding of GAN IF pathology has eluded researchers for many years. In a recent study(1) we demonstrate that the normal function of gigaxonin is to regulate the degradation of IF proteins via the proteasome. Our findings present the first direct link between GAN mutations and IF pathology; moreover, given the importance of IF aggregations in a wide range of disease conditions, our findings could have wider ramifications. </p>","PeriodicalId":74639,"journal":{"name":"Rare diseases (Austin, Tex.)","volume":"1 ","pages":"e25378"},"PeriodicalIF":0.0000,"publicationDate":"2013-06-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.4161/rdis.25378","citationCount":"10","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Rare diseases (Austin, Tex.)","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.4161/rdis.25378","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2013/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 10
Abstract
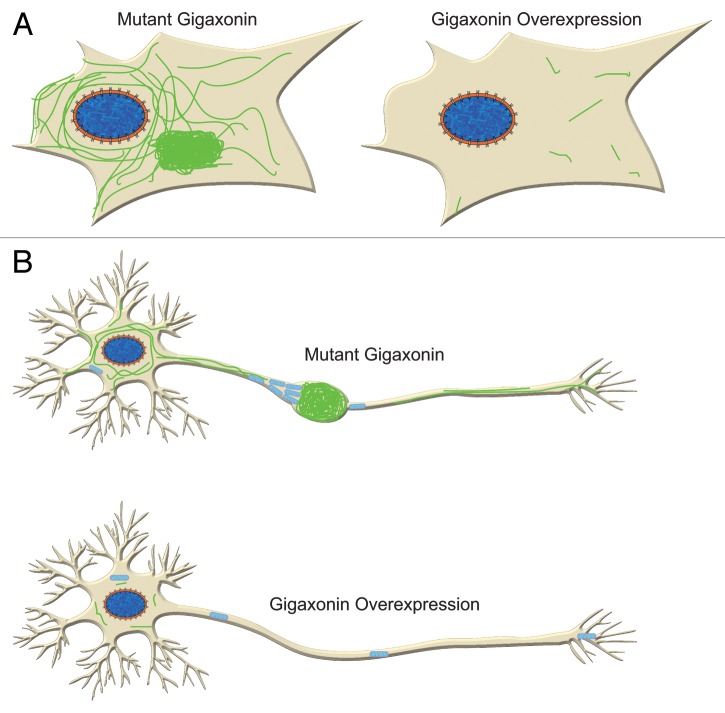
Giant axonal neuropathy (GAN)(1) is a rare autosomal recessive neurological disorder caused by mutations in the GAN gene that encodes gigaxonin, a member of the BTB/Kelch family of E3 ligase adaptor proteins.(1) This disease is characterized by the aggregation of Intermediate Filaments (IF)-cytoskeletal elements that play important roles in cell physiology including the regulation of cell shape, motility, mechanics and intra-cellular signaling. Although a range of cell types are affected in GAN, neurons display the most severe pathology, with neuronal intermediate filament accumulation and aggregation; this in turn causes axonal swellings or "giant axons." A mechanistic understanding of GAN IF pathology has eluded researchers for many years. In a recent study(1) we demonstrate that the normal function of gigaxonin is to regulate the degradation of IF proteins via the proteasome. Our findings present the first direct link between GAN mutations and IF pathology; moreover, given the importance of IF aggregations in a wide range of disease conditions, our findings could have wider ramifications.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


