Henrik H Kralund, Lilian Ousager, Nicolaas G Jaspers, Anja Raams, Erling B Pedersen, Else Gade, Anette Bygum
{"title":"Xeroderma Pigmentosum-Trichothiodystrophy overlap patient with novel XPD/ERCC2 mutation.","authors":"Henrik H Kralund, Lilian Ousager, Nicolaas G Jaspers, Anja Raams, Erling B Pedersen, Else Gade, Anette Bygum","doi":"10.4161/rdis.24932","DOIUrl":null,"url":null,"abstract":"<p><p>Xeroderma Pigmentosum (XP), Trichothiodystrophy (TTD) and Cockayne Syndrome (CS) are rare, recessive disorders caused by mutational defects in the Nucleotide Excision Repair (NER) pathway and/or disruption of basic cellular DNA transcription. To date, a multitude of mutations in the XPD/ERCC2 gene have been described, many of which give rise to NER- and DNA transcription related diseases, which share certain diagnostic features and few overlap patients have been described. Despite increasing understanding of the roles of XPD/ERCC2 in mammalian cells, there is still weak predictability of somatic outcome from many of these mutations. We demonstrate a patient, believed to represent an overlap between XP and TTD/CS. In addition to other organ dysfunctions, the young man presented with Photosensitivity, Ichthyosis, Brittle hair, Impaired physical and mental development, Decreased fertility and Short stature (PIBIDS) suggestive of TTD, but lacking the almost patognomonic \"tiger tail\" banding of the hair under polarized light. Additionally, he developed basal cell carcinoma aged 28, as well as adult onset kidney failure, features normally not associated with TTD but rather XP/CS. His freckled appearance also suggested XP, but fibroblast cultures only demonstrated x2 UV-sensitivity with expected NER and TFIIH-activity decrease. Genetic sequencing of the XPD/ERCC2 gene established the patient as heterozygote compound with a novel, N-terminal Y18H mutation and a known C-terminal (TTD) mutation, A725P. The possible interplay between gene products and the patient phenotype is discussed. </p>","PeriodicalId":74639,"journal":{"name":"Rare diseases (Austin, Tex.)","volume":"1 ","pages":"e24932"},"PeriodicalIF":0.0000,"publicationDate":"2013-05-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.4161/rdis.24932","citationCount":"12","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Rare diseases (Austin, Tex.)","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.4161/rdis.24932","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2013/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 12
Abstract
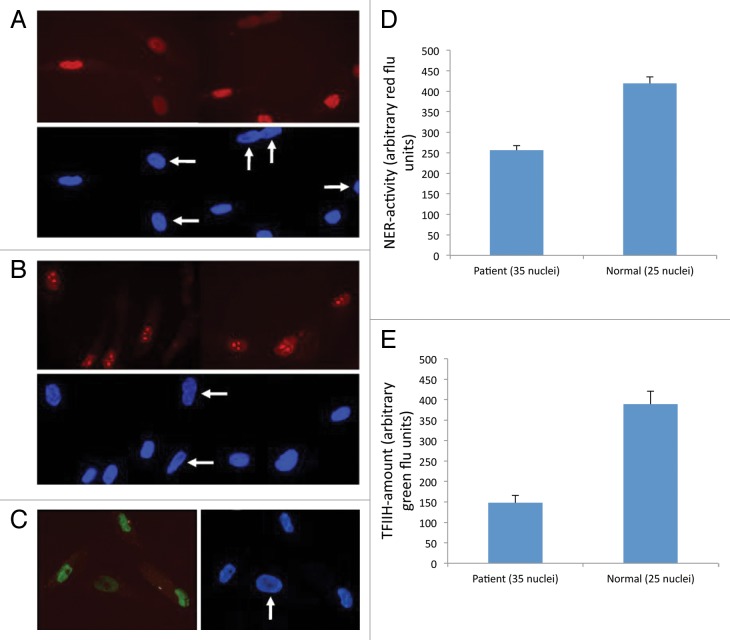
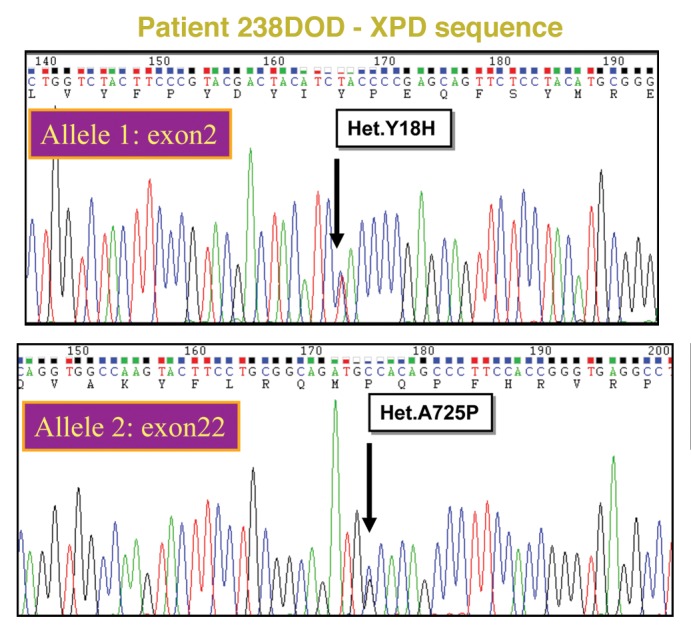
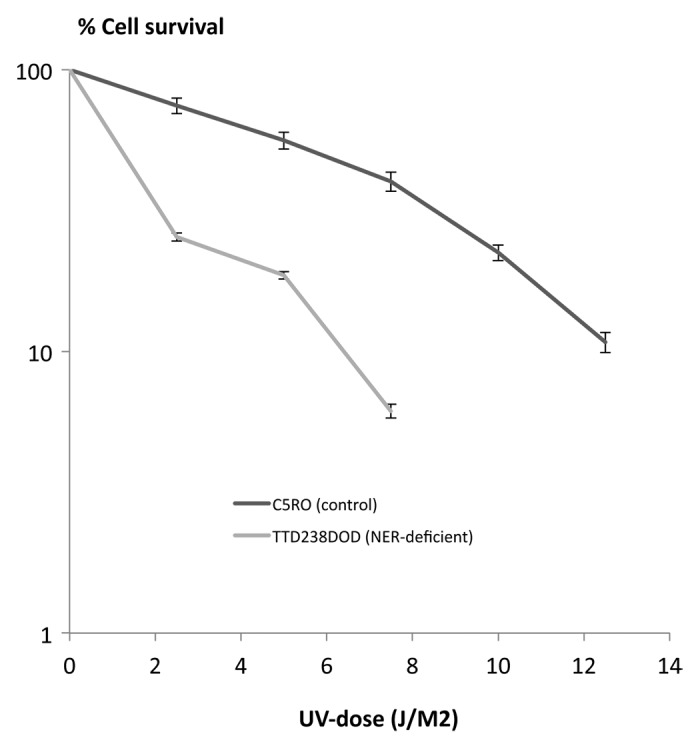
Xeroderma Pigmentosum (XP), Trichothiodystrophy (TTD) and Cockayne Syndrome (CS) are rare, recessive disorders caused by mutational defects in the Nucleotide Excision Repair (NER) pathway and/or disruption of basic cellular DNA transcription. To date, a multitude of mutations in the XPD/ERCC2 gene have been described, many of which give rise to NER- and DNA transcription related diseases, which share certain diagnostic features and few overlap patients have been described. Despite increasing understanding of the roles of XPD/ERCC2 in mammalian cells, there is still weak predictability of somatic outcome from many of these mutations. We demonstrate a patient, believed to represent an overlap between XP and TTD/CS. In addition to other organ dysfunctions, the young man presented with Photosensitivity, Ichthyosis, Brittle hair, Impaired physical and mental development, Decreased fertility and Short stature (PIBIDS) suggestive of TTD, but lacking the almost patognomonic "tiger tail" banding of the hair under polarized light. Additionally, he developed basal cell carcinoma aged 28, as well as adult onset kidney failure, features normally not associated with TTD but rather XP/CS. His freckled appearance also suggested XP, but fibroblast cultures only demonstrated x2 UV-sensitivity with expected NER and TFIIH-activity decrease. Genetic sequencing of the XPD/ERCC2 gene established the patient as heterozygote compound with a novel, N-terminal Y18H mutation and a known C-terminal (TTD) mutation, A725P. The possible interplay between gene products and the patient phenotype is discussed.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


