Hilde Vinje, Trygve Almøy, Kristian Hovde Liland, Lars Snipen
{"title":"A systematic search for discriminating sites in the 16S ribosomal RNA gene.","authors":"Hilde Vinje, Trygve Almøy, Kristian Hovde Liland, Lars Snipen","doi":"10.1186/2042-5783-4-2","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>The 16S rRNA is by far the most common genomic marker used for prokaryotic classification, and has been used extensively in metagenomic studies over recent years. Along the 16S gene there are regions with more or less variation across the kingdom of bacteria. Nine variable regions have been identified, flanked by more conserved parts of the sequence. It has been stated that the discriminatory power of the 16S marker lies in these variable regions. In the present study we wanted to examine this more closely, and used a supervised learning method to search systematically for sites that contribute to correct classification at either the phylum or genus level.</p><p><strong>Results: </strong>When classifying phyla the site selection algorithm located 50 discriminative sites. These were scattered over most of the alignments and only around half of them were located in the variable regions. The selected sites did, however, have an entropy significantly larger than expected, meaning they are sites of large variation. We found that the discriminative sites typically have a large entropy compared to their closest neighbours along the alignments. When classifying genera the site selection algorithm needed around 80% of the sites in the 16S gene before the classification error reached a minimum. This means that all variation, in both variable and conserved regions, is needed in order to separate genera.</p><p><strong>Conclusions: </strong>Our findings does not support the statement that the discriminative power of the 16S gene is located only in the variable regions. Variable regions are important, but just as many discriminative sites are found in the more conserved parts. The discriminative power is typically found in sites of large variation located inside shorter regions of higher conservation.</p>","PeriodicalId":18538,"journal":{"name":"Microbial Informatics and Experimentation","volume":"4 1","pages":"2"},"PeriodicalIF":0.0000,"publicationDate":"2014-01-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/2042-5783-4-2","citationCount":"21","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Microbial Informatics and Experimentation","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/2042-5783-4-2","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 21
Abstract
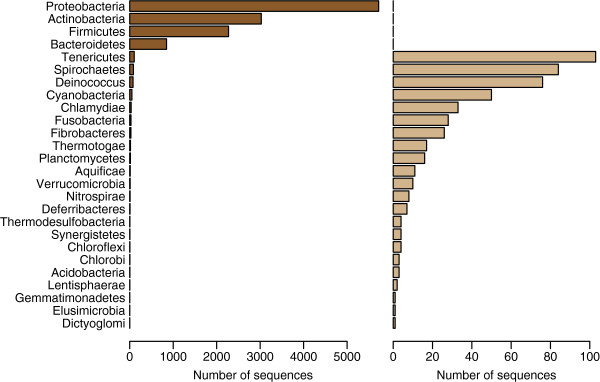
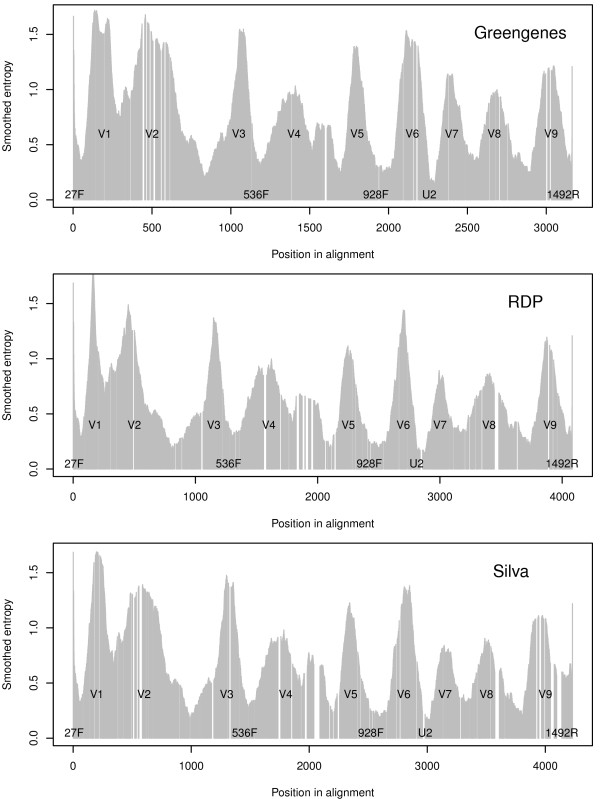
Background: The 16S rRNA is by far the most common genomic marker used for prokaryotic classification, and has been used extensively in metagenomic studies over recent years. Along the 16S gene there are regions with more or less variation across the kingdom of bacteria. Nine variable regions have been identified, flanked by more conserved parts of the sequence. It has been stated that the discriminatory power of the 16S marker lies in these variable regions. In the present study we wanted to examine this more closely, and used a supervised learning method to search systematically for sites that contribute to correct classification at either the phylum or genus level.
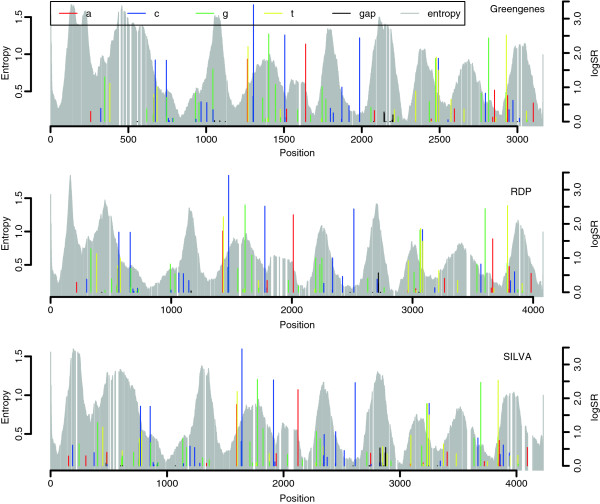
Results: When classifying phyla the site selection algorithm located 50 discriminative sites. These were scattered over most of the alignments and only around half of them were located in the variable regions. The selected sites did, however, have an entropy significantly larger than expected, meaning they are sites of large variation. We found that the discriminative sites typically have a large entropy compared to their closest neighbours along the alignments. When classifying genera the site selection algorithm needed around 80% of the sites in the 16S gene before the classification error reached a minimum. This means that all variation, in both variable and conserved regions, is needed in order to separate genera.
Conclusions: Our findings does not support the statement that the discriminative power of the 16S gene is located only in the variable regions. Variable regions are important, but just as many discriminative sites are found in the more conserved parts. The discriminative power is typically found in sites of large variation located inside shorter regions of higher conservation.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


