{"title":"Effective gene prediction by high resolution frequency estimator based on least-norm solution technique.","authors":"Manidipa Roy, Soma Barman","doi":"10.1186/1687-4153-2014-2","DOIUrl":null,"url":null,"abstract":"<p><p>Linear algebraic concept of subspace plays a significant role in the recent techniques of spectrum estimation. In this article, the authors have utilized the noise subspace concept for finding hidden periodicities in DNA sequence. With the vast growth of genomic sequences, the demand to identify accurately the protein-coding regions in DNA is increasingly rising. Several techniques of DNA feature extraction which involves various cross fields have come up in the recent past, among which application of digital signal processing tools is of prime importance. It is known that coding segments have a 3-base periodicity, while non-coding regions do not have this unique feature. One of the most important spectrum analysis techniques based on the concept of subspace is the least-norm method. The least-norm estimator developed in this paper shows sharp period-3 peaks in coding regions completely eliminating background noise. Comparison of proposed method with existing sliding discrete Fourier transform (SDFT) method popularly known as modified periodogram method has been drawn on several genes from various organisms and the results show that the proposed method has better as well as an effective approach towards gene prediction. Resolution, quality factor, sensitivity, specificity, miss rate, and wrong rate are used to establish superiority of least-norm gene prediction method over existing method. </p>","PeriodicalId":72957,"journal":{"name":"EURASIP journal on bioinformatics & systems biology","volume":"2014 1","pages":"2"},"PeriodicalIF":0.0000,"publicationDate":"2014-01-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3895782/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"EURASIP journal on bioinformatics & systems biology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/1687-4153-2014-2","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
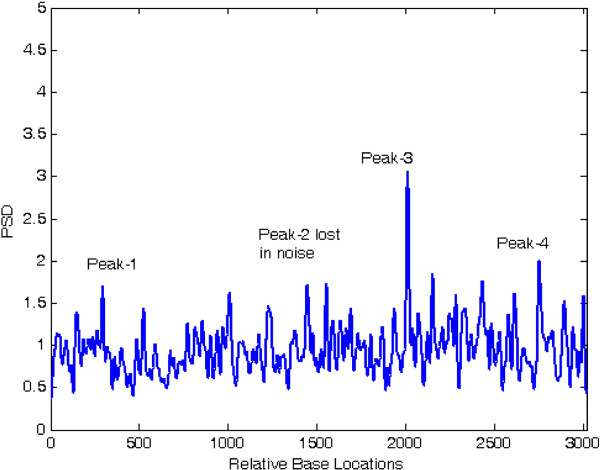
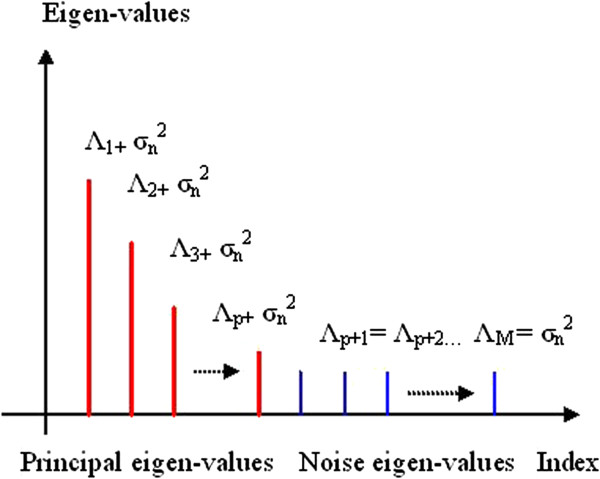
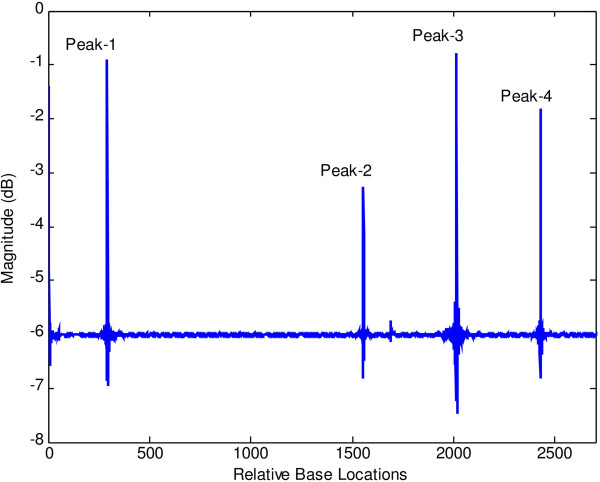
Linear algebraic concept of subspace plays a significant role in the recent techniques of spectrum estimation. In this article, the authors have utilized the noise subspace concept for finding hidden periodicities in DNA sequence. With the vast growth of genomic sequences, the demand to identify accurately the protein-coding regions in DNA is increasingly rising. Several techniques of DNA feature extraction which involves various cross fields have come up in the recent past, among which application of digital signal processing tools is of prime importance. It is known that coding segments have a 3-base periodicity, while non-coding regions do not have this unique feature. One of the most important spectrum analysis techniques based on the concept of subspace is the least-norm method. The least-norm estimator developed in this paper shows sharp period-3 peaks in coding regions completely eliminating background noise. Comparison of proposed method with existing sliding discrete Fourier transform (SDFT) method popularly known as modified periodogram method has been drawn on several genes from various organisms and the results show that the proposed method has better as well as an effective approach towards gene prediction. Resolution, quality factor, sensitivity, specificity, miss rate, and wrong rate are used to establish superiority of least-norm gene prediction method over existing method.
子空间的线性代数概念在最近的频谱估计技术中发挥了重要作用。在本文中,作者利用噪声子空间概念来寻找 DNA 序列中隐藏的周期性。随着基因组序列的大量增加,准确识别 DNA 中蛋白质编码区的需求也日益高涨。最近出现了几种涉及不同交叉领域的 DNA 特征提取技术,其中最重要的是数字信号处理工具的应用。众所周知,编码区段具有 3 个碱基的周期性,而非编码区段则没有这一独特特征。基于子空间概念的最重要频谱分析技术之一是最小正值法。本文开发的最小正估计器在编码区域显示出尖锐的 3 基周期峰,完全消除了背景噪声。通过对来自不同生物体的多个基因进行比较,将本文提出的方法与现有的滑动离散傅里叶变换(SDFT)方法(俗称修正周期图法)进行了比较,结果表明本文提出的方法在基因预测方面具有更好的效果。分辨率、品质因数、灵敏度、特异性、失误率和错误率被用来确定最小正态基因预测方法优于现有方法。

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


