Margaret Dellett, Kathleen Ann O'Hagan, Hilary Ann Alexandra Colyer, Ken I Mills
{"title":"Identification of gene networks associated with acute myeloid leukemia by comparative molecular methylation and expression profiling.","authors":"Margaret Dellett, Kathleen Ann O'Hagan, Hilary Ann Alexandra Colyer, Ken I Mills","doi":"10.4137/BIC.S3185","DOIUrl":null,"url":null,"abstract":"<p><p>Around 80% of acute myeloid leukemia (AML) patients achieve a complete remission, however many will relapse and ultimately die of their disease. The association between karyotype and prognosis has been studied extensively and identified patient cohorts as having favourable [e.g. t(8; 21), inv (16)/t(16; 16), t(15; 17)], intermediate [e.g. cytogenetically normal (NK-AML)] or adverse risk [e.g. complex karyotypes]. Previous studies have shown that gene expression profiling signatures can classify the sub-types of AML, although few reports have shown a similar feature by using methylation markers. The global methylation patterns in 19 diagnostic AML samples were investigated using the Methylated CpG Island Amplification Microarray (MCAM) method and CpG island microarrays containing 12,000 CpG sites. The first analysis, comparing favourable and intermediate cytogenetic risk groups, revealed significantly differentially methylated CpG sites (594 CpG islands) between the two subgroups. Mutations in the NPM1 gene occur at a high frequency (40%) within the NK-AML subgroup and are associated with a more favourable prognosis in these patients. A second analysis comparing the NPM1 mutant and wild-type research study subjects again identified distinct methylation profiles between these two subgroups. Network and pathway analysis revealed possible molecular mechanisms associated with the different risk and/or mutation sub-groups. This may result in a better classification of the risk groups, improved monitoring targets, or the identification of novel molecular therapies. </p>","PeriodicalId":72377,"journal":{"name":"Biomarkers in cancer","volume":"2 ","pages":"43-55"},"PeriodicalIF":0.0000,"publicationDate":"2010-03-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.4137/BIC.S3185","citationCount":"5","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biomarkers in cancer","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.4137/BIC.S3185","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2010/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 5
Abstract
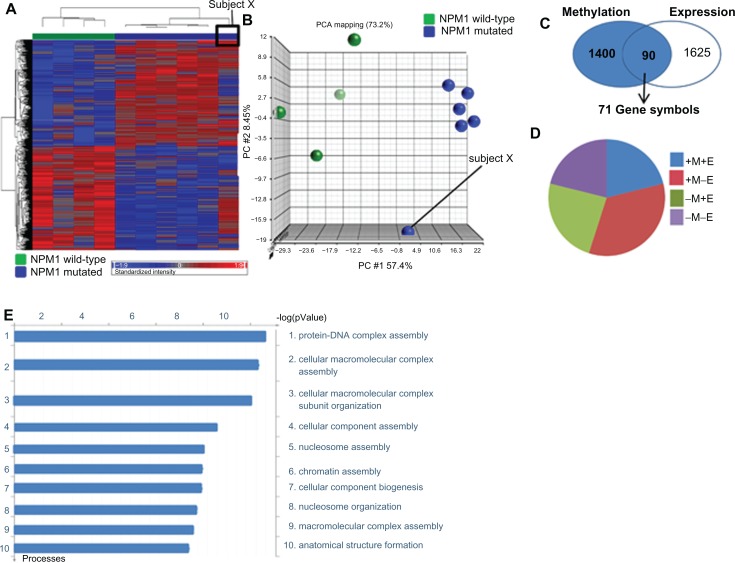
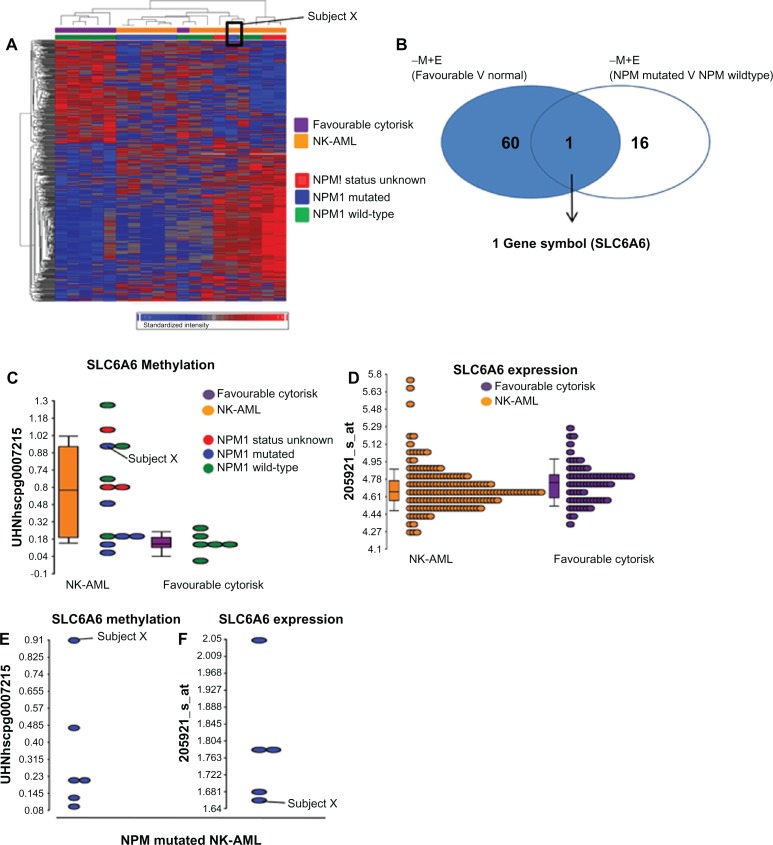
Around 80% of acute myeloid leukemia (AML) patients achieve a complete remission, however many will relapse and ultimately die of their disease. The association between karyotype and prognosis has been studied extensively and identified patient cohorts as having favourable [e.g. t(8; 21), inv (16)/t(16; 16), t(15; 17)], intermediate [e.g. cytogenetically normal (NK-AML)] or adverse risk [e.g. complex karyotypes]. Previous studies have shown that gene expression profiling signatures can classify the sub-types of AML, although few reports have shown a similar feature by using methylation markers. The global methylation patterns in 19 diagnostic AML samples were investigated using the Methylated CpG Island Amplification Microarray (MCAM) method and CpG island microarrays containing 12,000 CpG sites. The first analysis, comparing favourable and intermediate cytogenetic risk groups, revealed significantly differentially methylated CpG sites (594 CpG islands) between the two subgroups. Mutations in the NPM1 gene occur at a high frequency (40%) within the NK-AML subgroup and are associated with a more favourable prognosis in these patients. A second analysis comparing the NPM1 mutant and wild-type research study subjects again identified distinct methylation profiles between these two subgroups. Network and pathway analysis revealed possible molecular mechanisms associated with the different risk and/or mutation sub-groups. This may result in a better classification of the risk groups, improved monitoring targets, or the identification of novel molecular therapies.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


