{"title":"Identification of thresholds for dichotomizing DNA methylation data.","authors":"Yihua Liu, Yuan Ji, Peng Qiu","doi":"10.1186/1687-4153-2013-8","DOIUrl":null,"url":null,"abstract":"<p><p>: DNA methylation plays an important role in many biological processes by regulating gene expression. It is commonly accepted that turning on the DNA methylation leads to silencing of the expression of the corresponding genes. While methylation is often described as a binary on-off signal, it is typically measured using beta values derived from either microarray or sequencing technologies, which takes continuous values between 0 and 1. If we would like to interpret methylation in a binary fashion, appropriate thresholds are needed to dichotomize the continuous measurements. In this paper, we use data from The Cancer Genome Atlas project. For a total of 992 samples across five cancer types, both methylation and gene expression data are available. A bivariate extension of the StepMiner algorithm is used to identify thresholds for dichotomizing both methylation and expression data. Hypergeometric test is applied to identify CpG sites whose methylation status is significantly associated to silencing of the expression of their corresponding genes. The test is performed on either all five cancer types together or individual cancer types separately. We notice that the appropriate thresholds vary across different CpG sites. In addition, the negative association between methylation and expression is highly tissue specific.</p>","PeriodicalId":72957,"journal":{"name":"EURASIP journal on bioinformatics & systems biology","volume":"2013 1","pages":"8"},"PeriodicalIF":0.0000,"publicationDate":"2013-06-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/1687-4153-2013-8","citationCount":"12","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"EURASIP journal on bioinformatics & systems biology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/1687-4153-2013-8","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 12
Abstract
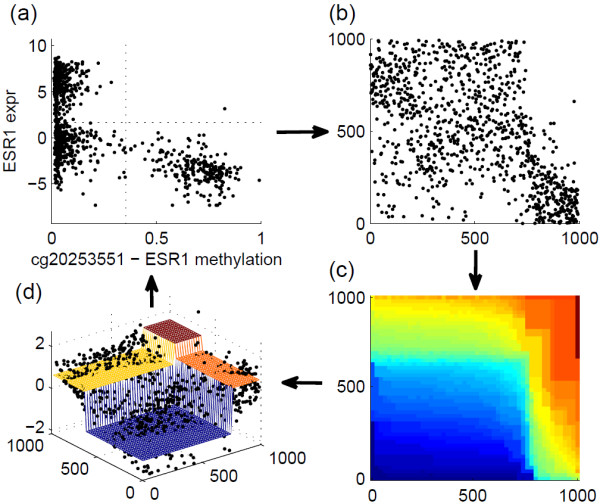
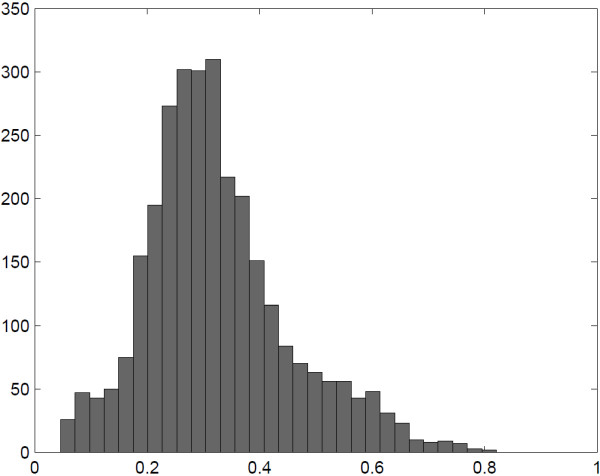
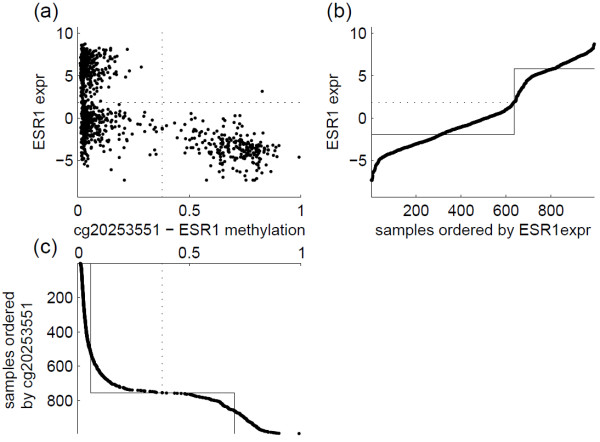
: DNA methylation plays an important role in many biological processes by regulating gene expression. It is commonly accepted that turning on the DNA methylation leads to silencing of the expression of the corresponding genes. While methylation is often described as a binary on-off signal, it is typically measured using beta values derived from either microarray or sequencing technologies, which takes continuous values between 0 and 1. If we would like to interpret methylation in a binary fashion, appropriate thresholds are needed to dichotomize the continuous measurements. In this paper, we use data from The Cancer Genome Atlas project. For a total of 992 samples across five cancer types, both methylation and gene expression data are available. A bivariate extension of the StepMiner algorithm is used to identify thresholds for dichotomizing both methylation and expression data. Hypergeometric test is applied to identify CpG sites whose methylation status is significantly associated to silencing of the expression of their corresponding genes. The test is performed on either all five cancer types together or individual cancer types separately. We notice that the appropriate thresholds vary across different CpG sites. In addition, the negative association between methylation and expression is highly tissue specific.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


