Zhiyun Cao, Sachin Yende, John A Kellum, Renã A S Robinson
{"title":"Additions to the Human Plasma Proteome via a Tandem MARS Depletion iTRAQ-Based Workflow.","authors":"Zhiyun Cao, Sachin Yende, John A Kellum, Renã A S Robinson","doi":"10.1155/2013/654356","DOIUrl":null,"url":null,"abstract":"<p><p>Robust platforms for determining differentially expressed proteins in biomarker and discovery studies using human plasma are of great interest. While increased depth in proteome coverage is desirable, it is associated with costs of experimental time due to necessary sample fractionation. We evaluated a robust quantitative proteomics workflow for its ability (1) to provide increased depth in plasma proteome coverage and (2) to give statistical insight useful for establishing differentially expressed plasma proteins. The workflow involves dual-stage immunodepletion on a multiple affinity removal system (MARS) column, iTRAQ tagging, offline strong-cation exchange chromatography, and liquid chromatography tandem mass spectrometry (LC-MS/MS). Independent workflow experiments were performed in triplicate on four plasma samples tagged with iTRAQ 4-plex reagents. After stringent criteria were applied to database searched results, 689 proteins with at least two spectral counts (SC) were identified. Depth in proteome coverage was assessed by comparison to the 2010 Human Plasma Proteome Reference Database in which our studies reveal 399 additional proteins which have not been previously reported. Additionally, we report on the technical variation of this quantitative workflow which ranges from ±11 to 30%.</p>","PeriodicalId":73474,"journal":{"name":"International journal of proteomics","volume":"2013 ","pages":"654356"},"PeriodicalIF":0.0000,"publicationDate":"2013-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1155/2013/654356","citationCount":"24","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"International journal of proteomics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2013/654356","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2013/2/19 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 24
Abstract
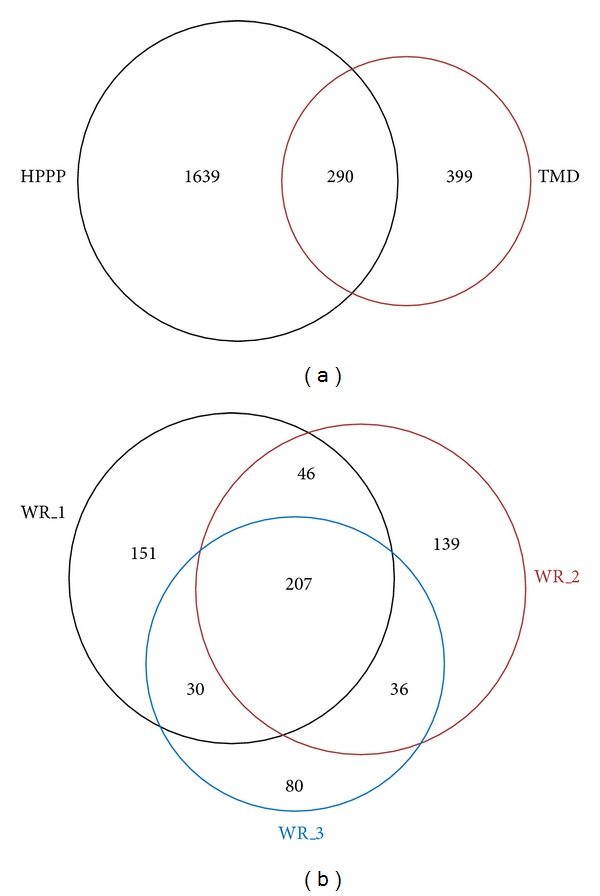
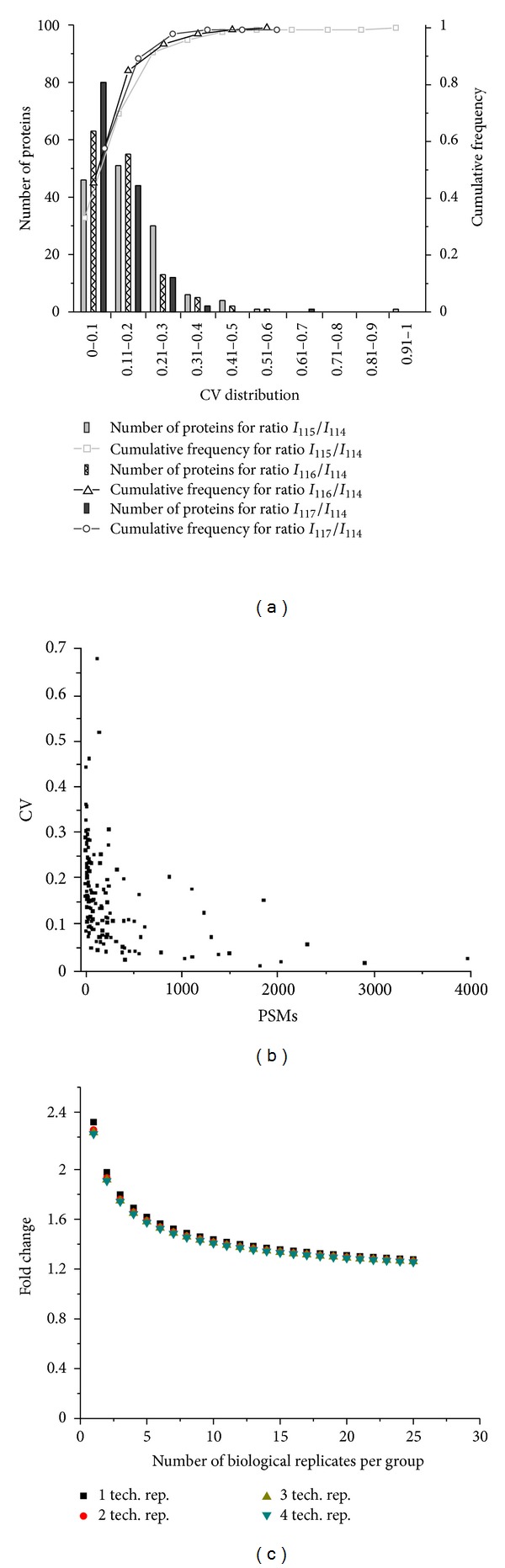
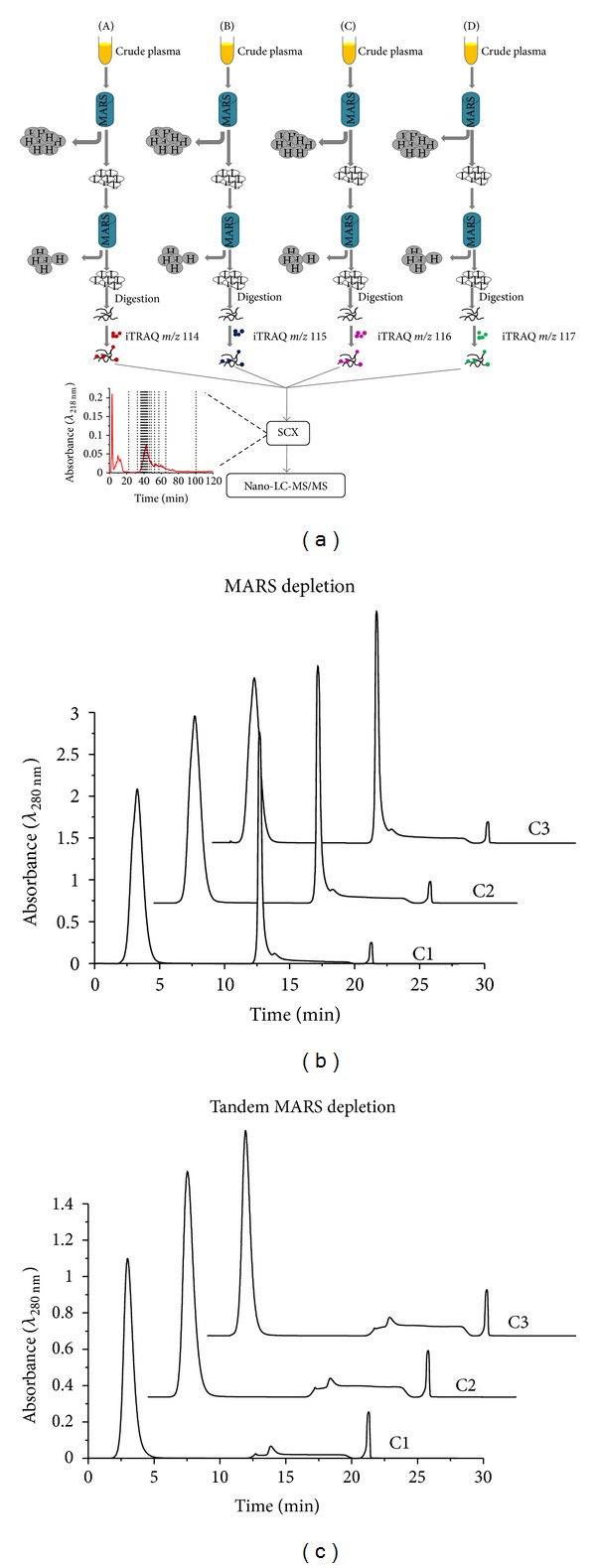
Robust platforms for determining differentially expressed proteins in biomarker and discovery studies using human plasma are of great interest. While increased depth in proteome coverage is desirable, it is associated with costs of experimental time due to necessary sample fractionation. We evaluated a robust quantitative proteomics workflow for its ability (1) to provide increased depth in plasma proteome coverage and (2) to give statistical insight useful for establishing differentially expressed plasma proteins. The workflow involves dual-stage immunodepletion on a multiple affinity removal system (MARS) column, iTRAQ tagging, offline strong-cation exchange chromatography, and liquid chromatography tandem mass spectrometry (LC-MS/MS). Independent workflow experiments were performed in triplicate on four plasma samples tagged with iTRAQ 4-plex reagents. After stringent criteria were applied to database searched results, 689 proteins with at least two spectral counts (SC) were identified. Depth in proteome coverage was assessed by comparison to the 2010 Human Plasma Proteome Reference Database in which our studies reveal 399 additional proteins which have not been previously reported. Additionally, we report on the technical variation of this quantitative workflow which ranges from ±11 to 30%.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


