David Murrugarra, Alan Veliz-Cuba, Boris Aguilar, Seda Arat, Reinhard Laubenbacher
{"title":"Modeling stochasticity and variability in gene regulatory networks.","authors":"David Murrugarra, Alan Veliz-Cuba, Boris Aguilar, Seda Arat, Reinhard Laubenbacher","doi":"10.1186/1687-4153-2012-5","DOIUrl":null,"url":null,"abstract":"<p><p> Modeling stochasticity in gene regulatory networks is an important and complex problem in molecular systems biology. To elucidate intrinsic noise, several modeling strategies such as the Gillespie algorithm have been used successfully. This article contributes an approach as an alternative to these classical settings. Within the discrete paradigm, where genes, proteins, and other molecular components of gene regulatory networks are modeled as discrete variables and are assigned as logical rules describing their regulation through interactions with other components. Stochasticity is modeled at the biological function level under the assumption that even if the expression levels of the input nodes of an update rule guarantee activation or degradation there is a probability that the process will not occur due to stochastic effects. This approach allows a finer analysis of discrete models and provides a natural setup for cell population simulations to study cell-to-cell variability. We applied our methods to two of the most studied regulatory networks, the outcome of lambda phage infection of bacteria and the p53-mdm2 complex.</p>","PeriodicalId":72957,"journal":{"name":"EURASIP journal on bioinformatics & systems biology","volume":"2012 1","pages":"5"},"PeriodicalIF":0.0000,"publicationDate":"2012-06-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/1687-4153-2012-5","citationCount":"82","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"EURASIP journal on bioinformatics & systems biology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/1687-4153-2012-5","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 82
Abstract
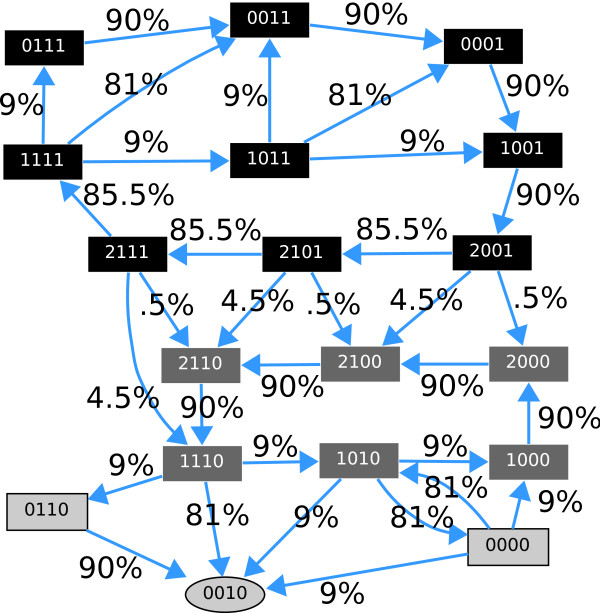
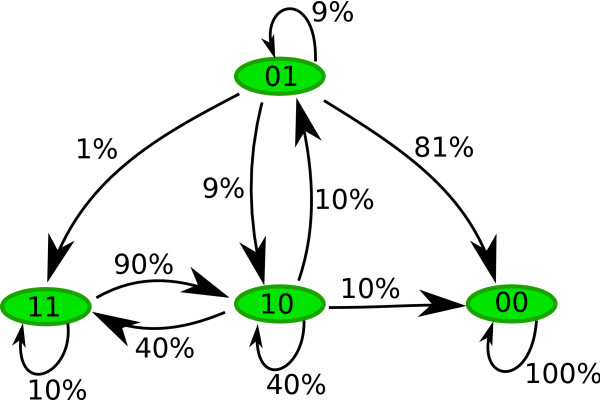
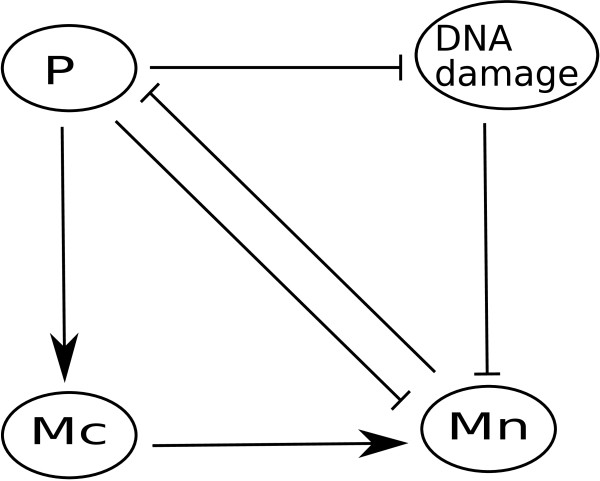
Modeling stochasticity in gene regulatory networks is an important and complex problem in molecular systems biology. To elucidate intrinsic noise, several modeling strategies such as the Gillespie algorithm have been used successfully. This article contributes an approach as an alternative to these classical settings. Within the discrete paradigm, where genes, proteins, and other molecular components of gene regulatory networks are modeled as discrete variables and are assigned as logical rules describing their regulation through interactions with other components. Stochasticity is modeled at the biological function level under the assumption that even if the expression levels of the input nodes of an update rule guarantee activation or degradation there is a probability that the process will not occur due to stochastic effects. This approach allows a finer analysis of discrete models and provides a natural setup for cell population simulations to study cell-to-cell variability. We applied our methods to two of the most studied regulatory networks, the outcome of lambda phage infection of bacteria and the p53-mdm2 complex.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


