{"title":"A network flow approach to predict drug targets from microarray data, disease genes and interactome network - case study on prostate cancer.","authors":"Shih-Heng Yeh, Hsiang-Yuan Yeh, Von-Wun Soo","doi":"10.1186/2043-9113-2-1","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Systematic approach for drug discovery is an emerging discipline in systems biology research area. It aims at integrating interaction data and experimental data to elucidate diseases and also raises new issues in drug discovery for cancer treatment. However, drug target discovery is still at a trial-and-error experimental stage and it is a challenging task to develop a prediction model that can systematically detect possible drug targets to deal with complex diseases.</p><p><strong>Methods: </strong>We integrate gene expression, disease genes and interaction networks to identify the effective drug targets which have a strong influence on disease genes using network flow approach. In the experiments, we adopt the microarray dataset containing 62 prostate cancer samples and 41 normal samples, 108 known prostate cancer genes and 322 approved drug targets treated in human extracted from DrugBank database to be candidate proteins as our test data. Using our method, we prioritize the candidate proteins and validate them to the known prostate cancer drug targets.</p><p><strong>Results: </strong>We successfully identify potential drug targets which are strongly related to the well known drugs for prostate cancer treatment and also discover more potential drug targets which raise the attention to biologists at present. We denote that it is hard to discover drug targets based only on differential expression changes due to the fact that those genes used to be drug targets may not always have significant expression changes. Comparing to previous methods that depend on the network topology attributes, they turn out that the genes having potential as drug targets are weakly correlated to critical points in a network. In comparison with previous methods, our results have highest mean average precision and also rank the position of the truly drug targets higher. It thereby verifies the effectiveness of our method.</p><p><strong>Conclusions: </strong>Our method does not know the real ideal routes in the disease network but it tries to find the feasible flow to give a strong influence to the disease genes through possible paths. We successfully formulate the identification of drug target prediction as a maximum flow problem on biological networks and discover potential drug targets in an accurate manner.</p>","PeriodicalId":73663,"journal":{"name":"Journal of clinical bioinformatics","volume":"2 1","pages":"1"},"PeriodicalIF":0.0000,"publicationDate":"2012-01-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3285036/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of clinical bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/2043-9113-2-1","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
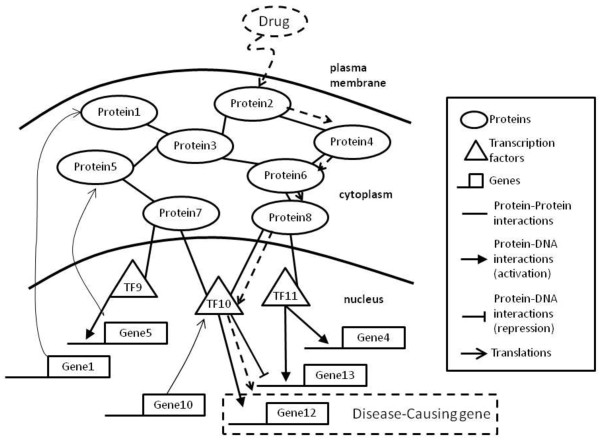
Background: Systematic approach for drug discovery is an emerging discipline in systems biology research area. It aims at integrating interaction data and experimental data to elucidate diseases and also raises new issues in drug discovery for cancer treatment. However, drug target discovery is still at a trial-and-error experimental stage and it is a challenging task to develop a prediction model that can systematically detect possible drug targets to deal with complex diseases.
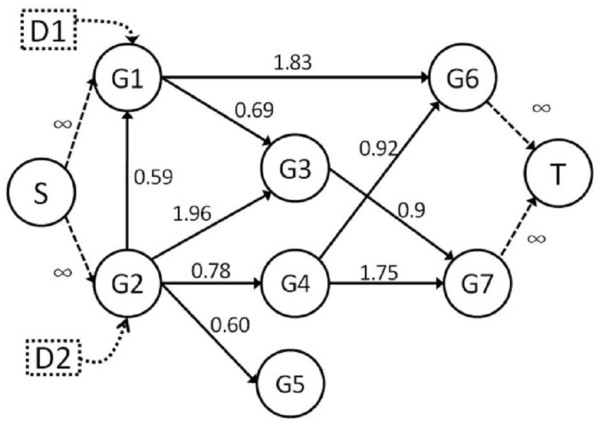
Methods: We integrate gene expression, disease genes and interaction networks to identify the effective drug targets which have a strong influence on disease genes using network flow approach. In the experiments, we adopt the microarray dataset containing 62 prostate cancer samples and 41 normal samples, 108 known prostate cancer genes and 322 approved drug targets treated in human extracted from DrugBank database to be candidate proteins as our test data. Using our method, we prioritize the candidate proteins and validate them to the known prostate cancer drug targets.
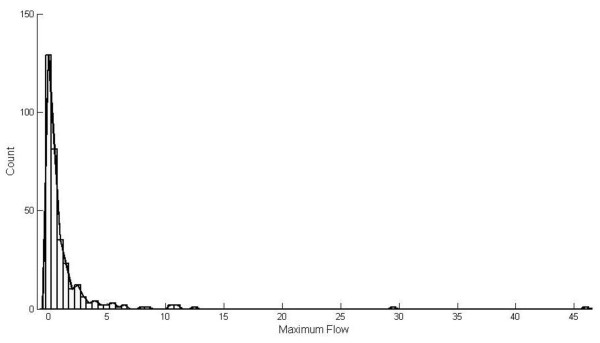
Results: We successfully identify potential drug targets which are strongly related to the well known drugs for prostate cancer treatment and also discover more potential drug targets which raise the attention to biologists at present. We denote that it is hard to discover drug targets based only on differential expression changes due to the fact that those genes used to be drug targets may not always have significant expression changes. Comparing to previous methods that depend on the network topology attributes, they turn out that the genes having potential as drug targets are weakly correlated to critical points in a network. In comparison with previous methods, our results have highest mean average precision and also rank the position of the truly drug targets higher. It thereby verifies the effectiveness of our method.
Conclusions: Our method does not know the real ideal routes in the disease network but it tries to find the feasible flow to give a strong influence to the disease genes through possible paths. We successfully formulate the identification of drug target prediction as a maximum flow problem on biological networks and discover potential drug targets in an accurate manner.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


