Mehdi Sadeghi, Hamid Pezeshk, Changiz Eslahchi, Sara Ahmadian, Sepideh Mah Abadi
{"title":"Construction of random perfect phylogeny matrix.","authors":"Mehdi Sadeghi, Hamid Pezeshk, Changiz Eslahchi, Sara Ahmadian, Sepideh Mah Abadi","doi":"10.2147/AABC.S13397","DOIUrl":null,"url":null,"abstract":"<p><strong>Purpose: </strong>Interest in developing methods appropriate for mapping increasing amounts of genome-wide molecular data are increasing rapidly. There is also an increasing need for methods that are able to efficiently simulate such data.</p><p><strong>Patients and methods: </strong>In this article, we provide a graph-theory approach to find the necessary and sufficient conditions for the existence of a phylogeny matrix with k nonidentical haplotypes, n single nucleotide polymorphisms (SNPs), and a population size of m for which the minimum allele frequency of each SNP is between two specific numbers a and b.</p><p><strong>Results: </strong>We introduce an O(max(n(2), nm)) algorithm for the random construction of such a phylogeny matrix. The running time of any algorithm for solving this problem would be Ω (nm).</p><p><strong>Conclusion: </strong>We have developed software, RAPPER, based on this algorithm, which is available at http://bioinf.cs.ipm.ir/softwares/RAPPER.</p>","PeriodicalId":53584,"journal":{"name":"Advances and Applications in Bioinformatics and Chemistry","volume":"3 ","pages":"89-96"},"PeriodicalIF":0.0000,"publicationDate":"2010-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.2147/AABC.S13397","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Advances and Applications in Bioinformatics and Chemistry","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.2147/AABC.S13397","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2010/11/16 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 0
Abstract
Purpose: Interest in developing methods appropriate for mapping increasing amounts of genome-wide molecular data are increasing rapidly. There is also an increasing need for methods that are able to efficiently simulate such data.
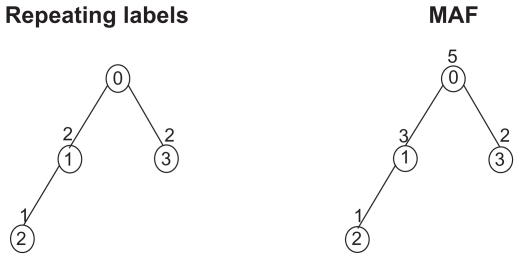
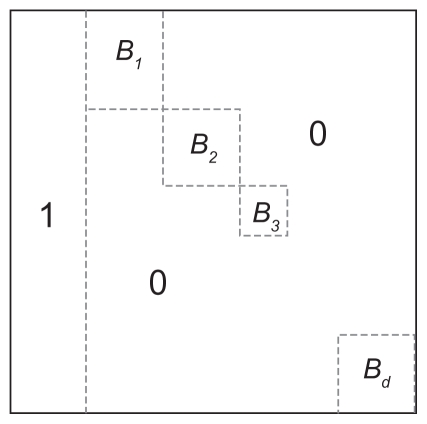
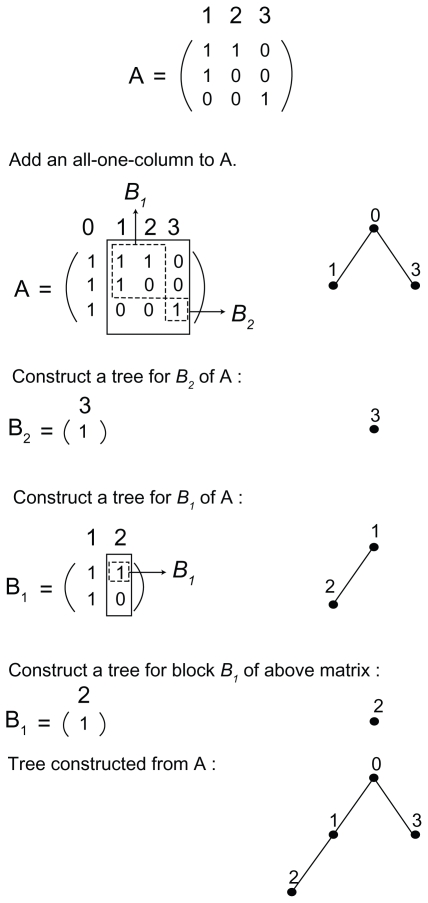
Patients and methods: In this article, we provide a graph-theory approach to find the necessary and sufficient conditions for the existence of a phylogeny matrix with k nonidentical haplotypes, n single nucleotide polymorphisms (SNPs), and a population size of m for which the minimum allele frequency of each SNP is between two specific numbers a and b.
Results: We introduce an O(max(n(2), nm)) algorithm for the random construction of such a phylogeny matrix. The running time of any algorithm for solving this problem would be Ω (nm).
Conclusion: We have developed software, RAPPER, based on this algorithm, which is available at http://bioinf.cs.ipm.ir/softwares/RAPPER.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


