Kelly L Mueller, Zeng-Quan Yang, Ramsi Haddad, Stephen P Ethier, Julie L Boerner
{"title":"EGFR/Met association regulates EGFR TKI resistance in breast cancer.","authors":"Kelly L Mueller, Zeng-Quan Yang, Ramsi Haddad, Stephen P Ethier, Julie L Boerner","doi":"10.1186/1750-2187-5-8","DOIUrl":null,"url":null,"abstract":"<p><p> Breast cancers show a lack of response to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs), despite 30% of tumors expressing EGFR. The mechanism of this resistance is unknown; however, we have recently shown that Met kinase activity compensates for loss of EGFR kinase activity in cell culture models. Met has been implicated in the pathogenesis of breast tumors and therefore may cooperate with EGFR for tumor growth. Here we have found that EGFR phosphorylation and cell proliferation is in part regulated by Met expression. In addition, we found that Met constitutive phosphorylation occurred independent of the Met ligand hepatocyte growth factor (HGF). Ligand-independent Met phosphorylation is mediated by Met amplification, mutation, or overexpression and by Met interaction with other cell surface molecules. In SUM229 breast cancer cells, we found that Met was not amplified or mutated, however it was overexpressed. Met overexpression did not directly correlate with ligand-independent Met phosphorylation as the SUM229 cell line was the only Met expressing breast cancer line with constitutive Met phosphorylation. Interestingly, Met expression did correlate with EGFR expression and we identified an EGFR/Met complex via co-immunoprecipitation. However, we only observed Met constitutive phosphorylation when c-Src also was part of this complex. Ligand-independent phosphorylation of Met was decreased by down regulating EGFR expression or by inhibiting c-Src kinase activity. Lastly, inhibiting EGFR and Met kinase activities resulted in a synergistic decrease in cell proliferation, supporting the idea that EGFR and Met functionally, as well as physically interact in breast cancer cells to regulate response to EGFR inhibitors.</p>","PeriodicalId":35051,"journal":{"name":"Journal of Molecular Signaling","volume":"5 ","pages":"8"},"PeriodicalIF":0.0000,"publicationDate":"2010-07-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/1750-2187-5-8","citationCount":"91","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Signaling","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/1750-2187-5-8","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 91
Abstract
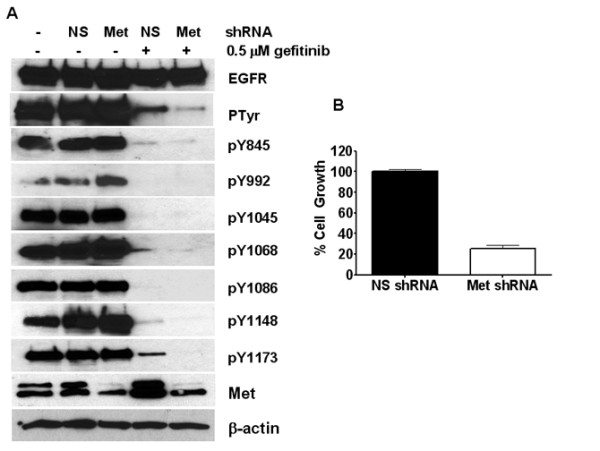
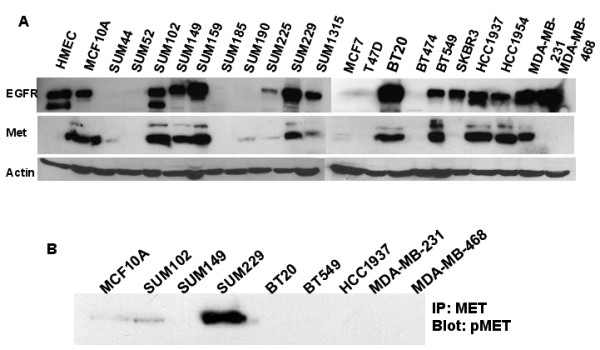
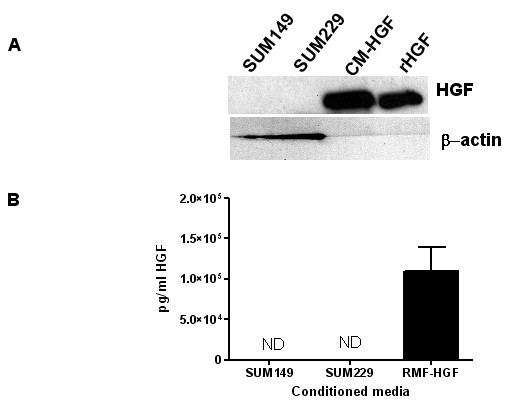
Breast cancers show a lack of response to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs), despite 30% of tumors expressing EGFR. The mechanism of this resistance is unknown; however, we have recently shown that Met kinase activity compensates for loss of EGFR kinase activity in cell culture models. Met has been implicated in the pathogenesis of breast tumors and therefore may cooperate with EGFR for tumor growth. Here we have found that EGFR phosphorylation and cell proliferation is in part regulated by Met expression. In addition, we found that Met constitutive phosphorylation occurred independent of the Met ligand hepatocyte growth factor (HGF). Ligand-independent Met phosphorylation is mediated by Met amplification, mutation, or overexpression and by Met interaction with other cell surface molecules. In SUM229 breast cancer cells, we found that Met was not amplified or mutated, however it was overexpressed. Met overexpression did not directly correlate with ligand-independent Met phosphorylation as the SUM229 cell line was the only Met expressing breast cancer line with constitutive Met phosphorylation. Interestingly, Met expression did correlate with EGFR expression and we identified an EGFR/Met complex via co-immunoprecipitation. However, we only observed Met constitutive phosphorylation when c-Src also was part of this complex. Ligand-independent phosphorylation of Met was decreased by down regulating EGFR expression or by inhibiting c-Src kinase activity. Lastly, inhibiting EGFR and Met kinase activities resulted in a synergistic decrease in cell proliferation, supporting the idea that EGFR and Met functionally, as well as physically interact in breast cancer cells to regulate response to EGFR inhibitors.
期刊介绍:
Journal of Molecular Signaling is an open access, peer-reviewed online journal that encompasses all aspects of molecular signaling. Molecular signaling is an exponentially growing field that encompasses different molecular aspects of cell signaling underlying normal and pathological conditions. Specifically, the research area of the journal is on the normal or aberrant molecular mechanisms involving receptors, G-proteins, kinases, phosphatases, and transcription factors in regulating cell proliferation, differentiation, apoptosis, and oncogenesis in mammalian cells. This area also covers the genetic and epigenetic changes that modulate the signaling properties of cells and the resultant physiological conditions.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


