Veronika М. Zainullina , Мichael А. Korotin , Victor L. Kozhevnikov
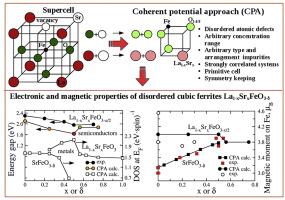
{"title":"Electronic properties of disordered perovskite-like ferrites: Coherent potential approach","authors":"Veronika М. Zainullina , Мichael А. Korotin , Victor L. Kozhevnikov","doi":"10.1016/j.progsolidstchem.2020.100284","DOIUrl":null,"url":null,"abstract":"<div><p>During the past decade, <em>ab initio</em><span> electronic structure methods have been extensively developed and employed for properties analysis of perovskites ABO</span><sub>3–δ</sub>, where A is a large cation and B is typically a 3<em>d</em><span><span><span> metal cation of smaller size. The perovskite structure is capable to withstand ample cation substitutions in both A and B sub-lattices and to simultaneously accommodate large amount of </span>oxygen vacancies (δ). The cation and anion defects result in considerable changes in </span>electronic spectrum<span> features and ensuing properties. In the variety of electronic structure calculation methods, the coherent potential approximation (CPA) is a special approach for studies of systems with disordered defects. The method is designed in order to overcome a number of restrictions that arise at employment of supercells such as defect ordering, limitations for defect types and concentrations, a drastic increase in calculation time with defect concentration, etc. The recently developed implementation of the CPA can be used for calculations of electronic spectrum and properties of solid state systems, including strongly correlated ones with an arbitrary concentration, arrangement and type of atomic structural defects. In this brief review, we consider the capabilities and restrictions of classical CPA-combined methods and represent a novel CPA methodology for the case study of electronic spectra and magnetic moments in several perovskite related disordered ferrites including SrFeO</span></span><sub>2.5</sub>, SrFeO<sub>3−δ</sub> and solid solutions La<sub>1−x</sub>Sr<sub>x</sub>FeO<sub>3−δ</sub><span>. These complex oxides with strong electronic correlations attract attention as inexpensive, environmentally friendly and robust materials for applications in high-temperature redox technologies, fuel cells, self-cleaning photocatalysis, water splitting, hydrogen and solar power engineering.</span></p></div>","PeriodicalId":415,"journal":{"name":"Progress in Solid State Chemistry","volume":"60 ","pages":"Article 100284"},"PeriodicalIF":10.5000,"publicationDate":"2020-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1016/j.progsolidstchem.2020.100284","citationCount":"4","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Progress in Solid State Chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0079678620300170","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, INORGANIC & NUCLEAR","Score":null,"Total":0}
引用次数: 4
Abstract
During the past decade, ab initio electronic structure methods have been extensively developed and employed for properties analysis of perovskites ABO3–δ, where A is a large cation and B is typically a 3d metal cation of smaller size. The perovskite structure is capable to withstand ample cation substitutions in both A and B sub-lattices and to simultaneously accommodate large amount of oxygen vacancies (δ). The cation and anion defects result in considerable changes in electronic spectrum features and ensuing properties. In the variety of electronic structure calculation methods, the coherent potential approximation (CPA) is a special approach for studies of systems with disordered defects. The method is designed in order to overcome a number of restrictions that arise at employment of supercells such as defect ordering, limitations for defect types and concentrations, a drastic increase in calculation time with defect concentration, etc. The recently developed implementation of the CPA can be used for calculations of electronic spectrum and properties of solid state systems, including strongly correlated ones with an arbitrary concentration, arrangement and type of atomic structural defects. In this brief review, we consider the capabilities and restrictions of classical CPA-combined methods and represent a novel CPA methodology for the case study of electronic spectra and magnetic moments in several perovskite related disordered ferrites including SrFeO2.5, SrFeO3−δ and solid solutions La1−xSrxFeO3−δ. These complex oxides with strong electronic correlations attract attention as inexpensive, environmentally friendly and robust materials for applications in high-temperature redox technologies, fuel cells, self-cleaning photocatalysis, water splitting, hydrogen and solar power engineering.
期刊介绍:
Progress in Solid State Chemistry offers critical reviews and specialized articles written by leading experts in the field, providing a comprehensive view of solid-state chemistry. It addresses the challenge of dispersed literature by offering up-to-date assessments of research progress and recent developments. Emphasis is placed on the relationship between physical properties and structural chemistry, particularly imperfections like vacancies and dislocations. The reviews published in Progress in Solid State Chemistry emphasize critical evaluation of the field, along with indications of current problems and future directions. Papers are not intended to be bibliographic in nature but rather to inform a broad range of readers in an inherently multidisciplinary field by providing expert treatises oriented both towards specialists in different areas of the solid state and towards nonspecialists. The authorship is international, and the subject matter will be of interest to chemists, materials scientists, physicists, metallurgists, crystallographers, ceramists, and engineers interested in the solid state.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


