Midbrain atrophy related to parkinsonism in a non-coding repeat expansion disorder: five cases of spinocerebellar ataxia type 31 with nigrostriatal dopaminergic dysfunction.
{"title":"Midbrain atrophy related to parkinsonism in a non-coding repeat expansion disorder: five cases of spinocerebellar ataxia type 31 with nigrostriatal dopaminergic dysfunction.","authors":"Ryohei Norioka, Keizo Sugaya, Aki Murayama, Tomoya Kawazoe, Shinsuke Tobisawa, Akihiro Kawata, Kazushi Takahashi","doi":"10.1186/s40673-021-00134-4","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Spinocerebellar ataxia type 31 (SCA31) is caused by non-coding pentanucleotide repeat expansions in the BEAN1 gene. Clinically, SCA31 is characterized by late adult-onset, pure cerebellar ataxia. To explore the association between parkinsonism and SCA31, five patients with SCA31 with concomitant nigrostriatal dopaminergic dysfunction (NSDD) development, including three cases of L-DOPA responsive parkinsonism, were analyzed.</p><p><strong>Methods: </strong>To assess regional brain atrophy, cross-sectional and longitudinal imaging analyses were retrospectively performed using magnetic resonance imaging (MRI) planimetry. The midbrain-to-pons (M/P) area ratio and cerebellar area were measured on midsagittal T1-weighted MRI in five patients with SCA31 with concomitant NSDD (NSDD(+)), 14 patients with SCA31 without NSDD (NSDD(-)), 32 patients with Parkinson's disease (PD), and 15 patients with progressive supranuclear palsy (PSP). Longitudinal changes in the M/P area ratio were assessed by serial MRI of NSDD(+) (n = 5) and NSDD(-) (n = 9).</p><p><strong>Results: </strong>The clinical characteristics assessed in the five patients with NSDD were as follows: the mean age at NSDD onset (72.0 ± 10.8 years), prominence of bradykinesia/akinesia (5/5), rigidity (4/5), tremor (2/5), dysautonomia (0/5), vertical gaze limitation (1/5), and abnormalities on <sup>123</sup>I-ioflupane dopamine transporter scintigraphy (3/3) and 3-Tesla neuromelanin MRI (4/4). A clear reduction in the midbrain area and the M/P area ratio was observed in the NSDD(+) group (p < 0.05) while there was no significant difference in disease duration or in the pons area among the NSDD(+), NSDD(-), and PD groups. There was also a significant difference in the midbrain and pons area between NSDD(+) and PSP (p < 0.05). Thus, mild but significant midbrain atrophy was observed in NSDD(+). A faster rate of decline in the midbrain area and the M/P area ratio was evident in NSDD(+) (p < 0.05).</p><p><strong>Conclusion: </strong>The clinical characteristics of the five patients with SCA31 with concomitant NSDD, together with the topographical pattern of atrophy, were inconsistent with PD, PSP, and multiple system atrophy, suggesting that SCA31 may manifest NSDD in association with the pathomechanisms underlying SCA31.</p>","PeriodicalId":36752,"journal":{"name":"Cerebellum and Ataxias","volume":" ","pages":"11"},"PeriodicalIF":0.0000,"publicationDate":"2021-03-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s40673-021-00134-4","citationCount":"4","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cerebellum and Ataxias","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s40673-021-00134-4","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"Medicine","Score":null,"Total":0}
引用次数: 4
Abstract
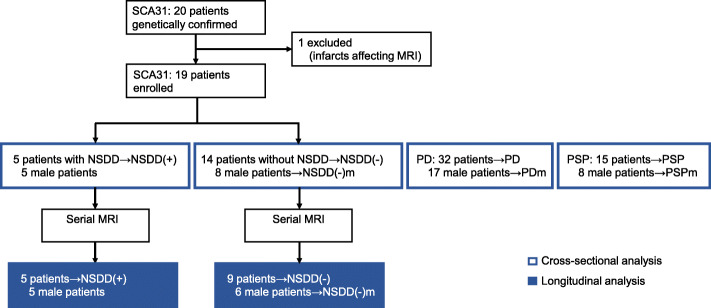
Background: Spinocerebellar ataxia type 31 (SCA31) is caused by non-coding pentanucleotide repeat expansions in the BEAN1 gene. Clinically, SCA31 is characterized by late adult-onset, pure cerebellar ataxia. To explore the association between parkinsonism and SCA31, five patients with SCA31 with concomitant nigrostriatal dopaminergic dysfunction (NSDD) development, including three cases of L-DOPA responsive parkinsonism, were analyzed.
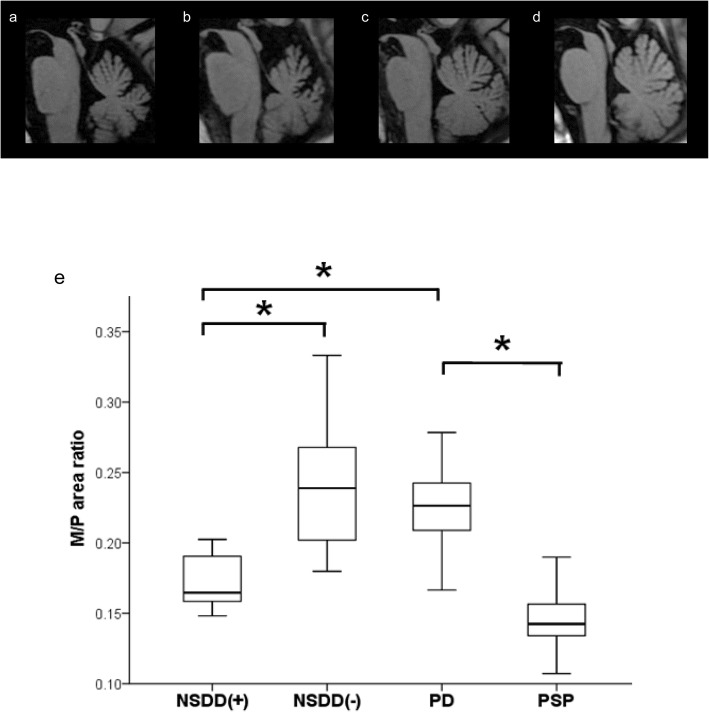
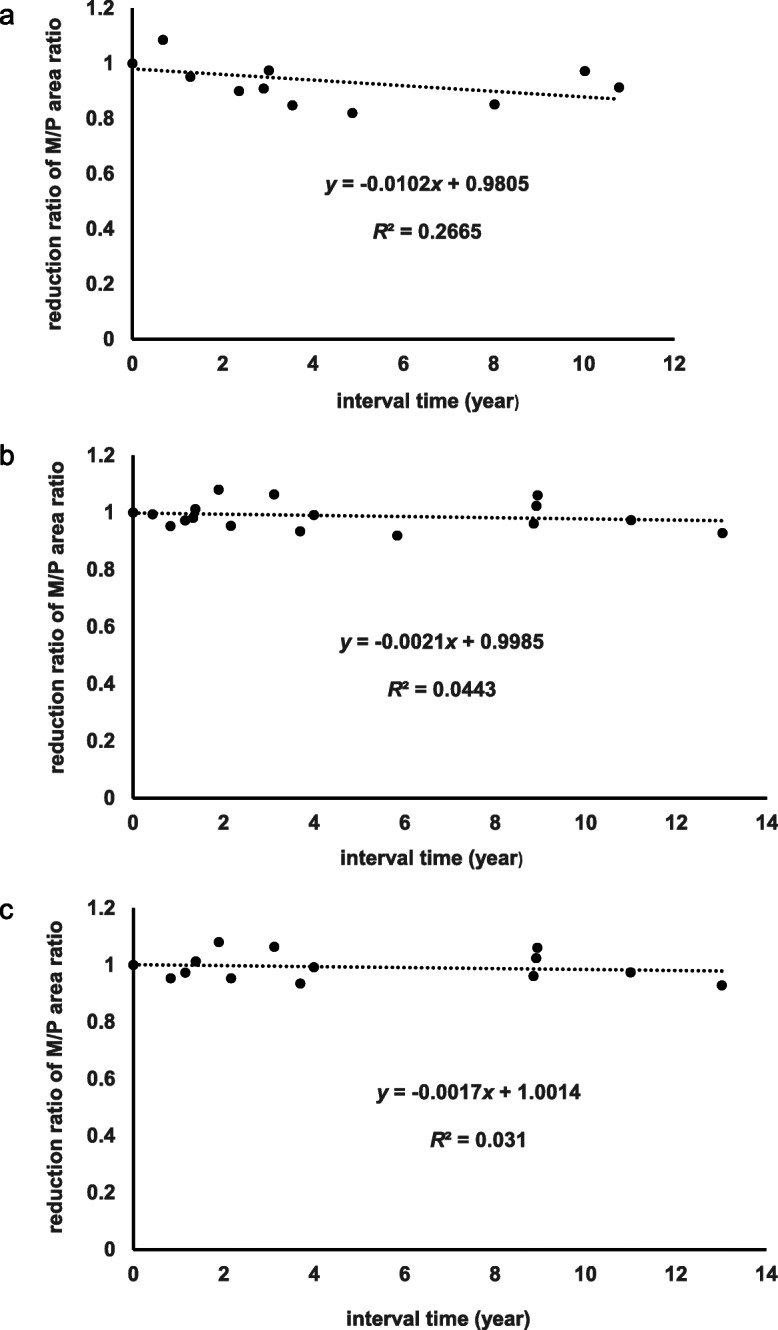
Methods: To assess regional brain atrophy, cross-sectional and longitudinal imaging analyses were retrospectively performed using magnetic resonance imaging (MRI) planimetry. The midbrain-to-pons (M/P) area ratio and cerebellar area were measured on midsagittal T1-weighted MRI in five patients with SCA31 with concomitant NSDD (NSDD(+)), 14 patients with SCA31 without NSDD (NSDD(-)), 32 patients with Parkinson's disease (PD), and 15 patients with progressive supranuclear palsy (PSP). Longitudinal changes in the M/P area ratio were assessed by serial MRI of NSDD(+) (n = 5) and NSDD(-) (n = 9).
Results: The clinical characteristics assessed in the five patients with NSDD were as follows: the mean age at NSDD onset (72.0 ± 10.8 years), prominence of bradykinesia/akinesia (5/5), rigidity (4/5), tremor (2/5), dysautonomia (0/5), vertical gaze limitation (1/5), and abnormalities on 123I-ioflupane dopamine transporter scintigraphy (3/3) and 3-Tesla neuromelanin MRI (4/4). A clear reduction in the midbrain area and the M/P area ratio was observed in the NSDD(+) group (p < 0.05) while there was no significant difference in disease duration or in the pons area among the NSDD(+), NSDD(-), and PD groups. There was also a significant difference in the midbrain and pons area between NSDD(+) and PSP (p < 0.05). Thus, mild but significant midbrain atrophy was observed in NSDD(+). A faster rate of decline in the midbrain area and the M/P area ratio was evident in NSDD(+) (p < 0.05).
Conclusion: The clinical characteristics of the five patients with SCA31 with concomitant NSDD, together with the topographical pattern of atrophy, were inconsistent with PD, PSP, and multiple system atrophy, suggesting that SCA31 may manifest NSDD in association with the pathomechanisms underlying SCA31.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


