Mohammad S Ghaemi, Adi L Tarca, Roberto Romero, Natalie Stanley, Ramin Fallahzadeh, Athena Tanada, Anthony Culos, Kazuo Ando, Xiaoyuan Han, Yair J Blumenfeld, Maurice L Druzin, Yasser Y El-Sayed, Ronald S Gibbs, Virginia D Winn, Kevin Contrepois, Xuefeng B Ling, Ronald J Wong, Gary M Shaw, David K Stevenson, Brice Gaudilliere, Nima Aghaeepour, Martin S Angst
{"title":"Proteomic signatures predict preeclampsia in individual cohorts but not across cohorts - implications for clinical biomarker studies.","authors":"Mohammad S Ghaemi, Adi L Tarca, Roberto Romero, Natalie Stanley, Ramin Fallahzadeh, Athena Tanada, Anthony Culos, Kazuo Ando, Xiaoyuan Han, Yair J Blumenfeld, Maurice L Druzin, Yasser Y El-Sayed, Ronald S Gibbs, Virginia D Winn, Kevin Contrepois, Xuefeng B Ling, Ronald J Wong, Gary M Shaw, David K Stevenson, Brice Gaudilliere, Nima Aghaeepour, Martin S Angst","doi":"10.1080/14767058.2021.1888915","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Early identification of pregnant women at risk for preeclampsia (PE) is important, as it will enable targeted interventions ahead of clinical manifestations. The quantitative analyses of plasma proteins feature prominently among molecular approaches used for risk prediction. However, derivation of protein signatures of sufficient predictive power has been challenging. The recent availability of platforms simultaneously assessing over 1000 plasma proteins offers broad examinations of the plasma proteome, which may enable the extraction of proteomic signatures with improved prognostic performance in prenatal care.</p><p><strong>Objective: </strong>The primary aim of this study was to examine the generalizability of proteomic signatures predictive of PE in two cohorts of pregnant women whose plasma proteome was interrogated with the same highly multiplexed platform. Establishing generalizability, or lack thereof, is critical to devise strategies facilitating the development of clinically useful predictive tests. A second aim was to examine the generalizability of protein signatures predictive of gestational age (GA) in uncomplicated pregnancies in the same cohorts to contrast physiological and pathological pregnancy outcomes.</p><p><strong>Study design: </strong>Serial blood samples were collected during the first, second, and third trimesters in 18 women who developed PE and 18 women with uncomplicated pregnancies (Stanford cohort). The second cohort (Detroit), used for comparative analysis, consisted of 76 women with PE and 90 women with uncomplicated pregnancies. Multivariate analyses were applied to infer predictive and cohort-specific proteomic models, which were then tested in the alternate cohort. Gene ontology (GO) analysis was performed to identify biological processes that were over-represented among top-ranked proteins associated with PE.</p><p><strong>Results: </strong>The model derived in the Stanford cohort was highly significant (<i>p</i> = 3.9E-15) and predictive (AUC = 0.96), but failed validation in the Detroit cohort (<i>p</i> = 9.7E-01, AUC = 0.50). Similarly, the model derived in the Detroit cohort was highly significant (<i>p</i> = 1.0E-21, AUC = 0.73), but failed validation in the Stanford cohort (<i>p</i> = 7.3E-02, AUC = 0.60). By contrast, proteomic models predicting GA were readily validated across the Stanford (<i>p</i> = 1.1E-454, <i>R</i> = 0.92) and Detroit cohorts (<i>p</i> = 1.1.E-92, <i>R</i> = 0.92) indicating that the proteomic assay performed well enough to infer a generalizable model across studied cohorts, which makes it less likely that technical aspects of the assay, including batch effects, accounted for observed differences.</p><p><strong>Conclusions: </strong>Results point to a broader issue relevant for proteomic and other omic discovery studies in patient cohorts suffering from a clinical syndrome, such as PE, driven by heterogeneous pathophysiologies. While novel technologies including highly multiplex proteomic arrays and adapted computational algorithms allow for novel discoveries for a particular study cohort, they may not readily generalize across cohorts. A likely reason is that the prevalence of pathophysiologic processes leading up to the \"same\" clinical syndrome can be distributed differently in different and smaller-sized cohorts. Signatures derived in individual cohorts may simply capture different facets of the spectrum of pathophysiologic processes driving a syndrome. Our findings have important implications for the design of omic studies of a syndrome like PE. They highlight the need for performing such studies in diverse and well-phenotyped patient populations that are large enough to characterize subsets of patients with shared pathophysiologies to then derive subset-specific signatures of sufficient predictive power.</p>","PeriodicalId":520807,"journal":{"name":"The journal of maternal-fetal & neonatal medicine : the official journal of the European Association of Perinatal Medicine, the Federation of Asia and Oceania Perinatal Societies, the International Society of Perinatal Obstetricians","volume":" ","pages":"5621-5628"},"PeriodicalIF":0.0000,"publicationDate":"2022-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1080/14767058.2021.1888915","citationCount":"13","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The journal of maternal-fetal & neonatal medicine : the official journal of the European Association of Perinatal Medicine, the Federation of Asia and Oceania Perinatal Societies, the International Society of Perinatal Obstetricians","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1080/14767058.2021.1888915","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2021/3/2 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 13
Abstract
Background: Early identification of pregnant women at risk for preeclampsia (PE) is important, as it will enable targeted interventions ahead of clinical manifestations. The quantitative analyses of plasma proteins feature prominently among molecular approaches used for risk prediction. However, derivation of protein signatures of sufficient predictive power has been challenging. The recent availability of platforms simultaneously assessing over 1000 plasma proteins offers broad examinations of the plasma proteome, which may enable the extraction of proteomic signatures with improved prognostic performance in prenatal care.
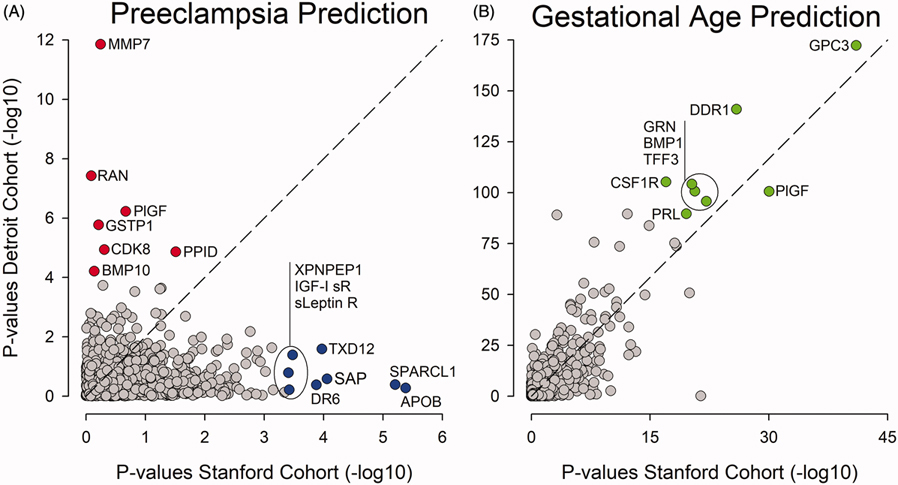
Objective: The primary aim of this study was to examine the generalizability of proteomic signatures predictive of PE in two cohorts of pregnant women whose plasma proteome was interrogated with the same highly multiplexed platform. Establishing generalizability, or lack thereof, is critical to devise strategies facilitating the development of clinically useful predictive tests. A second aim was to examine the generalizability of protein signatures predictive of gestational age (GA) in uncomplicated pregnancies in the same cohorts to contrast physiological and pathological pregnancy outcomes.
Study design: Serial blood samples were collected during the first, second, and third trimesters in 18 women who developed PE and 18 women with uncomplicated pregnancies (Stanford cohort). The second cohort (Detroit), used for comparative analysis, consisted of 76 women with PE and 90 women with uncomplicated pregnancies. Multivariate analyses were applied to infer predictive and cohort-specific proteomic models, which were then tested in the alternate cohort. Gene ontology (GO) analysis was performed to identify biological processes that were over-represented among top-ranked proteins associated with PE.
Results: The model derived in the Stanford cohort was highly significant (p = 3.9E-15) and predictive (AUC = 0.96), but failed validation in the Detroit cohort (p = 9.7E-01, AUC = 0.50). Similarly, the model derived in the Detroit cohort was highly significant (p = 1.0E-21, AUC = 0.73), but failed validation in the Stanford cohort (p = 7.3E-02, AUC = 0.60). By contrast, proteomic models predicting GA were readily validated across the Stanford (p = 1.1E-454, R = 0.92) and Detroit cohorts (p = 1.1.E-92, R = 0.92) indicating that the proteomic assay performed well enough to infer a generalizable model across studied cohorts, which makes it less likely that technical aspects of the assay, including batch effects, accounted for observed differences.
Conclusions: Results point to a broader issue relevant for proteomic and other omic discovery studies in patient cohorts suffering from a clinical syndrome, such as PE, driven by heterogeneous pathophysiologies. While novel technologies including highly multiplex proteomic arrays and adapted computational algorithms allow for novel discoveries for a particular study cohort, they may not readily generalize across cohorts. A likely reason is that the prevalence of pathophysiologic processes leading up to the "same" clinical syndrome can be distributed differently in different and smaller-sized cohorts. Signatures derived in individual cohorts may simply capture different facets of the spectrum of pathophysiologic processes driving a syndrome. Our findings have important implications for the design of omic studies of a syndrome like PE. They highlight the need for performing such studies in diverse and well-phenotyped patient populations that are large enough to characterize subsets of patients with shared pathophysiologies to then derive subset-specific signatures of sufficient predictive power.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


