Ismail Yaz, Sevil Oskay Halacli, Canberk Ipsir, Baris Ulum, Elif Soyak Aytekin, Hacer Neslihan Bildik, Melike Ocak, Hanife Avci, Fatma Visal Okur, Hayriye Hizarcioglu Gulsen, Hulya Demir, Ayse Metin, Alev Ozon, Baris Kuskonmaz, Ilhan Tezcan, Saliha Esenboga, Deniz Cagdas
{"title":"Beyond the Classical Triad: Atypical Presentations and Regulatory T Cell Phenotyping in a Cohort of IPEX Patients.","authors":"Ismail Yaz, Sevil Oskay Halacli, Canberk Ipsir, Baris Ulum, Elif Soyak Aytekin, Hacer Neslihan Bildik, Melike Ocak, Hanife Avci, Fatma Visal Okur, Hayriye Hizarcioglu Gulsen, Hulya Demir, Ayse Metin, Alev Ozon, Baris Kuskonmaz, Ilhan Tezcan, Saliha Esenboga, Deniz Cagdas","doi":"10.1007/s10875-025-01934-7","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Immune dysregulation, polyendocrinopathy, enteropathy, and X-linked(IPEX) syndrome caused by FOXP3 mutations is rare. FOXP3 is a transcription factor required for the regulatory T cell (Treg) development/function.</p><p><strong>Aim: </strong>We aimed to characterize the clinical, immunologic, and genetic features of a single-center cohort of IPEX syndrome.</p><p><strong>Patients and methods: </strong>We present the clinical/immunological/genetic features of 12 patients with IPEX syndrome. We used whole exome and Sanger sequencing for the diagnosis/familial segregation. We performed immunophenotyping and measured Treg percentage and FOXP3 expression in peripheral blood by flow cytometric analysis.</p><p><strong>Results: </strong>Median age at diagnosis was 2.5 years (range: 0.3-22 years). Common clinical manifestations were infections (n = 9, 75%), allergies (n = 8, 67%), autoimmunity (n = 7, 58%), enteropathy (n = 7, 58%), and lymphoproliferation (n = 3, 25%). Atypical initial presentations included class IV lupus nephritis, a SCID-like immunophenotype (CD3<sup>+</sup> T cells: 4% [100/µL]; CD4<sup>+</sup> T cells: 3%, CD8<sup>+</sup> T cells: 1%, CD19<sup>+</sup> B cells: 81%, CD16/56<sup>+</sup> NK cells: 13%), and isolated hypogammaglobulinemia persisting for years during follow-up. At the time of diagnosis, three (25%) patients had leukopenia, six (50%) had lymphopenia and two (17%) had neutropenia. Eosinophilia was observed in 42% of patients (25% mild, 17% moderate). Six different variants in FOXP3 were characterized in 12 patients from nine unrelated families. Four (33%) patients underwent hematopoietic stem cell transplantation (HSCT). Overall, three (25%) patients died due to infections. One patient died due to HSCT-related catheter complications, one patient died in an accident. Among the transplanted patients, two are alive and well. Among the non-transplanted patients, five are alive and are being followed up at our center. Treg (CD4<sup>+</sup>CD127<sup>-/low</sup>CD25<sup>+</sup>Foxp3<sup>+</sup>) percentage was low in eight patients compared to healthy controls (p < 0.001). FOXP3 expression was low in all the patients compared to healthy controls.</p><p><strong>Conclusion: </strong>Atypical presentations make the diagnosis of IPEX syndrome challenging. This study expands the current knowledge of IPEX syndrome by describing a single-center cohort with certain atypical manifestations and by confirming previously reported rare phenotypes. Elucidating the genetic basis of immunodeficiency diseases contributes to improving diagnostic approaches and patient management.</p>","PeriodicalId":15531,"journal":{"name":"Journal of Clinical Immunology","volume":"45 1","pages":"148"},"PeriodicalIF":5.7000,"publicationDate":"2025-10-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12540517/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Clinical Immunology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10875-025-01934-7","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"IMMUNOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Immune dysregulation, polyendocrinopathy, enteropathy, and X-linked(IPEX) syndrome caused by FOXP3 mutations is rare. FOXP3 is a transcription factor required for the regulatory T cell (Treg) development/function.
Aim: We aimed to characterize the clinical, immunologic, and genetic features of a single-center cohort of IPEX syndrome.
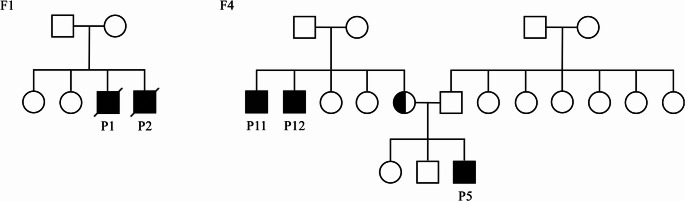
Patients and methods: We present the clinical/immunological/genetic features of 12 patients with IPEX syndrome. We used whole exome and Sanger sequencing for the diagnosis/familial segregation. We performed immunophenotyping and measured Treg percentage and FOXP3 expression in peripheral blood by flow cytometric analysis.
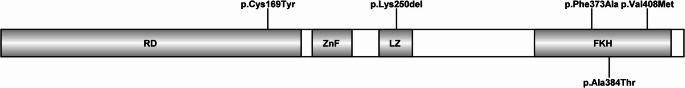
Results: Median age at diagnosis was 2.5 years (range: 0.3-22 years). Common clinical manifestations were infections (n = 9, 75%), allergies (n = 8, 67%), autoimmunity (n = 7, 58%), enteropathy (n = 7, 58%), and lymphoproliferation (n = 3, 25%). Atypical initial presentations included class IV lupus nephritis, a SCID-like immunophenotype (CD3+ T cells: 4% [100/µL]; CD4+ T cells: 3%, CD8+ T cells: 1%, CD19+ B cells: 81%, CD16/56+ NK cells: 13%), and isolated hypogammaglobulinemia persisting for years during follow-up. At the time of diagnosis, three (25%) patients had leukopenia, six (50%) had lymphopenia and two (17%) had neutropenia. Eosinophilia was observed in 42% of patients (25% mild, 17% moderate). Six different variants in FOXP3 were characterized in 12 patients from nine unrelated families. Four (33%) patients underwent hematopoietic stem cell transplantation (HSCT). Overall, three (25%) patients died due to infections. One patient died due to HSCT-related catheter complications, one patient died in an accident. Among the transplanted patients, two are alive and well. Among the non-transplanted patients, five are alive and are being followed up at our center. Treg (CD4+CD127-/lowCD25+Foxp3+) percentage was low in eight patients compared to healthy controls (p < 0.001). FOXP3 expression was low in all the patients compared to healthy controls.
Conclusion: Atypical presentations make the diagnosis of IPEX syndrome challenging. This study expands the current knowledge of IPEX syndrome by describing a single-center cohort with certain atypical manifestations and by confirming previously reported rare phenotypes. Elucidating the genetic basis of immunodeficiency diseases contributes to improving diagnostic approaches and patient management.
期刊介绍:
The Journal of Clinical Immunology publishes impactful papers in the realm of human immunology, delving into the diagnosis, pathogenesis, prognosis, or treatment of human diseases. The journal places particular emphasis on primary immunodeficiencies and related diseases, encompassing inborn errors of immunity in a broad sense, their underlying genotypes, and diverse phenotypes. These phenotypes include infection, malignancy, allergy, auto-inflammation, and autoimmunity. We welcome a broad spectrum of studies in this domain, spanning genetic discovery, clinical description, immunologic assessment, diagnostic approaches, prognosis evaluation, and treatment interventions. Case reports are considered if they are genuinely original and accompanied by a concise review of the relevant medical literature, illustrating how the novel case study advances the field. The instructions to authors provide detailed guidance on the four categories of papers accepted by the journal.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


