Lyna-Nour Hamidi, Jack Christopher Drda, Meriem Belhocine, Hannah-Laure Elfassy, Stéphanie Ducharme-Bénard, Maxime Chayer-Lanthier, Bushra Sultana, Sylvain Lanthier
{"title":"Case Report: Mevalonate kinase deficiency: an underdiagnosed cause of ischemic stroke-characterization of a novel genetic variant.","authors":"Lyna-Nour Hamidi, Jack Christopher Drda, Meriem Belhocine, Hannah-Laure Elfassy, Stéphanie Ducharme-Bénard, Maxime Chayer-Lanthier, Bushra Sultana, Sylvain Lanthier","doi":"10.3389/fimmu.2025.1651819","DOIUrl":null,"url":null,"abstract":"<p><p>Mevalonate kinase deficiency (MKD) is an inherited autoinflammatory syndrome resulting from impaired isoprenoid biosynthesis due to biallelic mevalonate kinase (<i>MVK</i>) mutations. This metabolic defect leads to dysregulated innate immunity, particularly excessive interleukin-1β release. While typically presenting in childhood with periodic fevers, expanding evidence links MKD to heterogeneous adult phenotypes with immune-mediated end-organ damage. We report an adult male presenting with leg pain and finger cyanosis followed by acute ischemic stroke, macular rash, and lymphadenopathies. He exhibited classical markers of innate immune activation, including persistent elevation of C-reactive protein. Genetic testing identified compound heterozygosity for the known <i>MVK</i> pathogenic variant c.1129G>A (V377I) and a novel missense variant, c.1049A>C (Q350P). Structural modeling of Q350P revealed disruption of the GHMP kinase domain, predicted to destabilize mevalonate kinase conformation and impair its function. The measurement of mevalonate kinase activity in lymphocytes was at 55% (normal >60%). Interleukin-1β blockade with canakinumab was initiated, and the blood markers of inflammation normalized, further supporting a central role for innate immune dysregulation. This case highlights a novel <i>MVK</i> missense variant (Q350P) with subnormal mevalonate kinase activity. The patient's compound heterozygous state with partially preserved mevalonate kinase activity may explain the attenuated systemic features and the delayed clinical onset. Remarkably, ischemic stroke was part of the initial presentation, suggesting that mevalonate kinase deficiency can manifest primarily through thrombo-inflammatory complications in adulthood, even in the absence of recurrent febrile episodes. This expands the phenotypic spectrum of MKD and underscores the need to consider adult-onset autoinflammatory syndromes in the differential diagnosis of cryptogenic ischemic strokes with markers of systemic inflammation. It also supports the utility of cytokine-targeted therapies in such contexts.</p>","PeriodicalId":12622,"journal":{"name":"Frontiers in Immunology","volume":"16 ","pages":"1651819"},"PeriodicalIF":5.9000,"publicationDate":"2025-10-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12531262/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Frontiers in Immunology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.3389/fimmu.2025.1651819","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q1","JCRName":"IMMUNOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
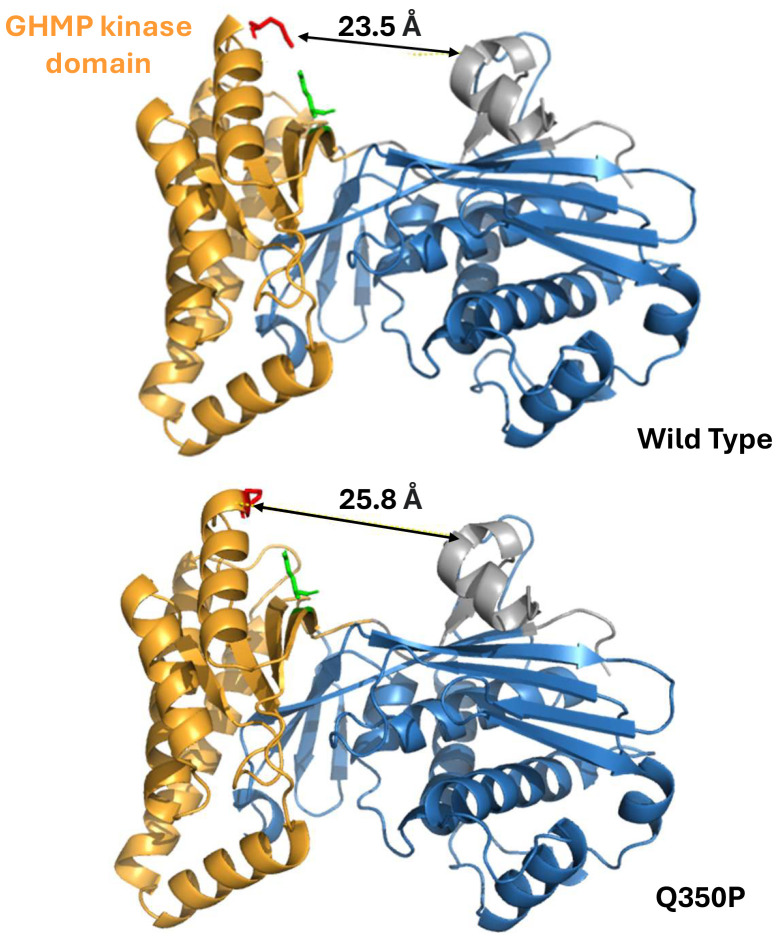
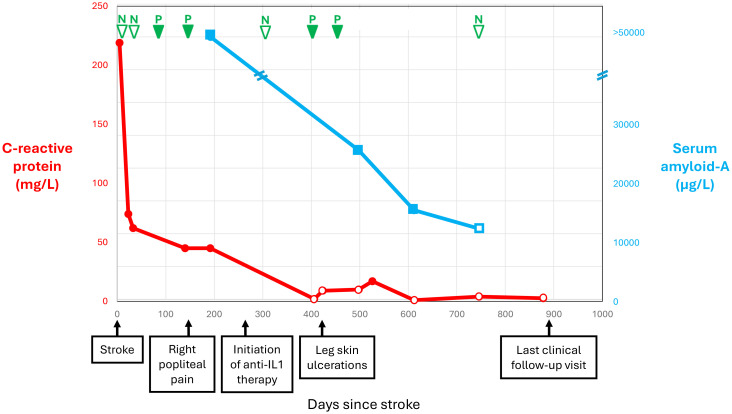
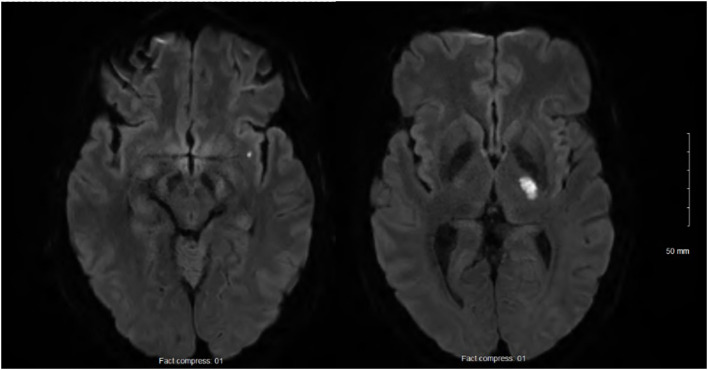
Mevalonate kinase deficiency (MKD) is an inherited autoinflammatory syndrome resulting from impaired isoprenoid biosynthesis due to biallelic mevalonate kinase (MVK) mutations. This metabolic defect leads to dysregulated innate immunity, particularly excessive interleukin-1β release. While typically presenting in childhood with periodic fevers, expanding evidence links MKD to heterogeneous adult phenotypes with immune-mediated end-organ damage. We report an adult male presenting with leg pain and finger cyanosis followed by acute ischemic stroke, macular rash, and lymphadenopathies. He exhibited classical markers of innate immune activation, including persistent elevation of C-reactive protein. Genetic testing identified compound heterozygosity for the known MVK pathogenic variant c.1129G>A (V377I) and a novel missense variant, c.1049A>C (Q350P). Structural modeling of Q350P revealed disruption of the GHMP kinase domain, predicted to destabilize mevalonate kinase conformation and impair its function. The measurement of mevalonate kinase activity in lymphocytes was at 55% (normal >60%). Interleukin-1β blockade with canakinumab was initiated, and the blood markers of inflammation normalized, further supporting a central role for innate immune dysregulation. This case highlights a novel MVK missense variant (Q350P) with subnormal mevalonate kinase activity. The patient's compound heterozygous state with partially preserved mevalonate kinase activity may explain the attenuated systemic features and the delayed clinical onset. Remarkably, ischemic stroke was part of the initial presentation, suggesting that mevalonate kinase deficiency can manifest primarily through thrombo-inflammatory complications in adulthood, even in the absence of recurrent febrile episodes. This expands the phenotypic spectrum of MKD and underscores the need to consider adult-onset autoinflammatory syndromes in the differential diagnosis of cryptogenic ischemic strokes with markers of systemic inflammation. It also supports the utility of cytokine-targeted therapies in such contexts.
期刊介绍:
Frontiers in Immunology is a leading journal in its field, publishing rigorously peer-reviewed research across basic, translational and clinical immunology. This multidisciplinary open-access journal is at the forefront of disseminating and communicating scientific knowledge and impactful discoveries to researchers, academics, clinicians and the public worldwide.
Frontiers in Immunology is the official Journal of the International Union of Immunological Societies (IUIS). Encompassing the entire field of Immunology, this journal welcomes papers that investigate basic mechanisms of immune system development and function, with a particular emphasis given to the description of the clinical and immunological phenotype of human immune disorders, and on the definition of their molecular basis.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


