Weak *CO-Binding Dopants in Cu-Based Single-Atom Alloys Serving as Diffusion-Assisting Mediators to Facilitate C–C Coupling
IF 3.2
3区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
Copper-based single-atom alloy (SAA) catalysts exhibit tunable C–C coupling behavior during CO2 reduction, governed by dopant-dependent metal–carbon (M–C) interactions. Combining density functional theory (DFT) calculation and ab initio molecular dynamics (AIMD) simulation, this study systematically investigates dopant effects across three C–C coupling pathways: *CO+*CO, *CO+*COH, and *CO+*CHO. Compared to *COH and *CHO, *CO is identified as the primary migratory intermediate governing coupling kinetics due to its weak adsorption and low coordination number. Strong M–C interactions (e.g., Ni-doped Cu) anchor *CO at dopant sites, suppressing migration and increasing C–C coupling barriers. Conversely, weak M–C interactions (e.g., Zn-doped Cu) destabilize *CO adsorption, enabling its migration between Cu sites and reducing C–C coupling energy barriers compared to pristine Cu. We propose an assisted-diffusion mechanism in which dopants with weak M–C interactions promote *CO migration for C–C coupling by acting as diffusion mediators rather than active adsorption sites, thereby enhancing the Faraday efficiency of the overall multicarbon product. These findings provide atomic-scale insights for designing high-activity Cu-based SAAs via the targeted modulation of dopant–C interactions.
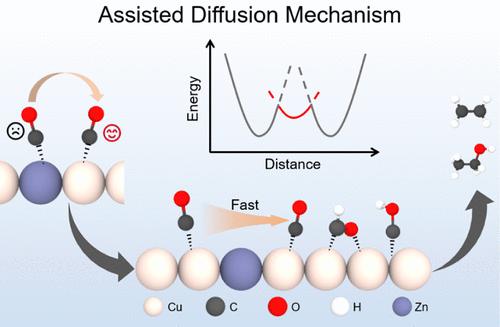
cu基单原子合金中的弱*共结合掺杂剂作为促进C-C耦合的扩散辅助介质
铜基单原子合金(SAA)催化剂在CO2还原过程中表现出可调节的C-C耦合行为,受掺杂剂依赖的金属-碳(M-C)相互作用的控制。本研究结合密度泛函理论(DFT)计算和从头算分子动力学(AIMD)模拟,系统地研究了*CO+*CO、*CO+*COH和*CO+*CHO三种C-C耦合途径中的掺杂效应。与*COH和*CHO相比,由于*CO吸附弱,配位数低,被认为是控制耦合动力学的主要迁移中间体。强M-C相互作用(如ni掺杂Cu)在掺杂位点锚定*CO,抑制迁移并增加C-C偶联屏障。相反,与原始Cu相比,弱M-C相互作用(例如,锌掺杂Cu)破坏了*CO吸附的稳定性,使其能够在Cu位点之间迁移,并降低了C-C耦合能垒。我们提出了一种辅助扩散机制,其中具有弱M-C相互作用的掺杂剂作为扩散介质而不是活性吸附位点促进*CO向C-C耦合的迁移,从而提高了整个多碳产物的法拉第效率。这些发现为通过靶向调制掺杂剂- c相互作用来设计高活性的基于cu的SAAs提供了原子尺度的见解。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

The Journal of Physical Chemistry C
化学-材料科学:综合
CiteScore
6.50
自引率
8.10%
发文量
2047
审稿时长
1.8 months
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


