{"title":"DCMF-PPI: a protein-protein interaction predictor based on dynamic condition and multi-feature fusion.","authors":"Siqi Chen, Anhong Zheng, Weichi Yu, Chao Zhan","doi":"10.1186/s12859-025-06272-4","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>The identification of protein-protein interaction (PPI) plays a crucial role in understanding the mechanisms of complex biological processes. Current research in predicting PPI has shown remarkable progress by integrating protein information with PPI topology structure. Nevertheless, these approaches frequently overlook the dynamic nature of protein and PPI structures during cellular processes, including conformational alterations and variations in binding affinities under diverse environmental circumstances. Additionally, the insufficient availability of comprehensive protein data hinders accurate protein representation. Consequently, these shortcomings restrict the model's generalizability and predictive precision.</p><p><strong>Results: </strong>To address this, we introduce DCMF-PPI (Dynamic condition and multi-feature fusion framework for PPI), a novel hybrid framework that integrates dynamic modeling, multi-scale feature extraction, and probabilistic graph representation learning. DCMF-PPI comprises three core modules: (1) PortT5-GAT Module: The protein language model PortT5 is utilized to extract residue-level protein features, which are integrated with dynamic temporal dependencies. Graph attention networks are then employed to capture context-aware structural variations in protein interactions; (2) MPSWA Module: Employs parallel convolutional neural networks combined with wavelet transform to extract multi-scale features from diverse protein residue types, enhancing the representation of sequence and structural heterogeneity; (3) VGAE Module: Utilizes a Variational Graph Autoencoder to learn probabilistic latent representations, facilitating dynamic modeling of PPI graph structures and capturing uncertainty in interaction dynamics.</p><p><strong>Conclusion: </strong>We conducted comprehensive experiments on benchmark datasets demonstrating that DCMF-PPI outperforms state-of-the-art methods in PPI prediction, achieving significant improvements in accuracy, precision, and recall. The framework's ability to fuse dynamic conditions and multi-level features highlights its effectiveness in modeling real-world biological complexities, positioning it as a robust tool for advancing PPI research and downstream applications in systems biology and drug discovery.</p>","PeriodicalId":8958,"journal":{"name":"BMC Bioinformatics","volume":"26 1","pages":"247"},"PeriodicalIF":3.3000,"publicationDate":"2025-10-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12522320/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s12859-025-06272-4","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
Background: The identification of protein-protein interaction (PPI) plays a crucial role in understanding the mechanisms of complex biological processes. Current research in predicting PPI has shown remarkable progress by integrating protein information with PPI topology structure. Nevertheless, these approaches frequently overlook the dynamic nature of protein and PPI structures during cellular processes, including conformational alterations and variations in binding affinities under diverse environmental circumstances. Additionally, the insufficient availability of comprehensive protein data hinders accurate protein representation. Consequently, these shortcomings restrict the model's generalizability and predictive precision.
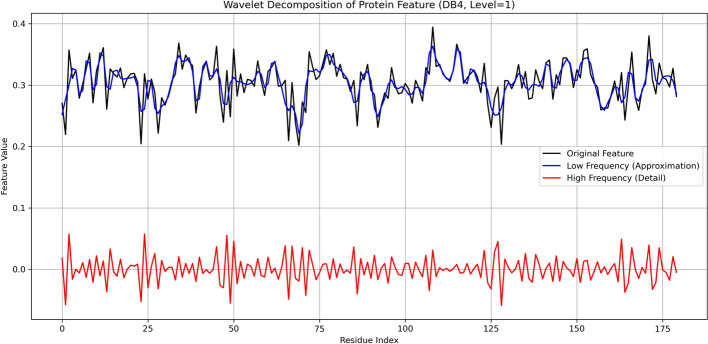
Results: To address this, we introduce DCMF-PPI (Dynamic condition and multi-feature fusion framework for PPI), a novel hybrid framework that integrates dynamic modeling, multi-scale feature extraction, and probabilistic graph representation learning. DCMF-PPI comprises three core modules: (1) PortT5-GAT Module: The protein language model PortT5 is utilized to extract residue-level protein features, which are integrated with dynamic temporal dependencies. Graph attention networks are then employed to capture context-aware structural variations in protein interactions; (2) MPSWA Module: Employs parallel convolutional neural networks combined with wavelet transform to extract multi-scale features from diverse protein residue types, enhancing the representation of sequence and structural heterogeneity; (3) VGAE Module: Utilizes a Variational Graph Autoencoder to learn probabilistic latent representations, facilitating dynamic modeling of PPI graph structures and capturing uncertainty in interaction dynamics.
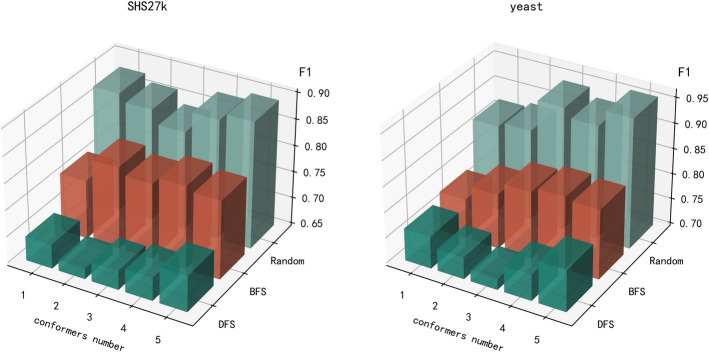
Conclusion: We conducted comprehensive experiments on benchmark datasets demonstrating that DCMF-PPI outperforms state-of-the-art methods in PPI prediction, achieving significant improvements in accuracy, precision, and recall. The framework's ability to fuse dynamic conditions and multi-level features highlights its effectiveness in modeling real-world biological complexities, positioning it as a robust tool for advancing PPI research and downstream applications in systems biology and drug discovery.
期刊介绍:
BMC Bioinformatics is an open access, peer-reviewed journal that considers articles on all aspects of the development, testing and novel application of computational and statistical methods for the modeling and analysis of all kinds of biological data, as well as other areas of computational biology.
BMC Bioinformatics is part of the BMC series which publishes subject-specific journals focused on the needs of individual research communities across all areas of biology and medicine. We offer an efficient, fair and friendly peer review service, and are committed to publishing all sound science, provided that there is some advance in knowledge presented by the work.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


