{"title":"Comparative quantum chemical analysis of dexamethasone and hydrocortisone: electronic structure, and reactivity indices using DFT","authors":"Masoumeh Eskandari-Nasab, Zainab Moosavi-Tekyeh, Mansoureh Zahedi-Tabrizi","doi":"10.1007/s00894-025-06522-5","DOIUrl":null,"url":null,"abstract":"<div><h3>Context</h3><p>The biological activity of steroidal compounds such as dexamethasone (DX) and hydrocortisone (HC) is closely linked to subtle variations in their molecular structure and electronic properties. This study provides a comparative quantum chemical analysis of DX and HC to clarify how these differences influence hydrogen bonding strength, reactivity, and their potential interactions with the glucocorticoid receptor (GR). Optimized geometries, natural bond orbital (NBO) analyses, frontier molecular orbitals (FMO), global reactivity descriptors, and average local ionization energy (ALIE) calculations demonstrate that DX exhibits greater polarity and electrophilic character compared to HC. These differences help explain the stronger receptor binding affinity observed for DX. Indeed, notably, despite the inherent limitations of gas-phase DFT calculations compared to experimental X-ray data, the theoretical results exhibit good agreement with experimental observations, suggesting the reliability of the computational approach in predicting molecular interactions within the GR active site.</p><h3>Methods</h3><p>All quantum chemical calculations were performed using density functional theory (DFT) with the B3LYP functional and 6-311++G(d,p) basis set. Structural optimization, FMO analysis, global reactivity descriptors, and dipole moment evaluations were carried out in Gaussian 09. NBO analysis was performed with NBO 5.0. Average local ionization energy (ALIE) surfaces were generated using Multiwfn 3.8, and molecular visualizations were produced with GaussView 5.0.</p></div>","PeriodicalId":651,"journal":{"name":"Journal of Molecular Modeling","volume":"31 11","pages":""},"PeriodicalIF":2.5000,"publicationDate":"2025-10-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Modeling","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s00894-025-06522-5","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Context
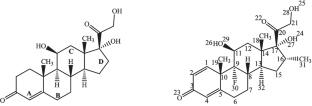
The biological activity of steroidal compounds such as dexamethasone (DX) and hydrocortisone (HC) is closely linked to subtle variations in their molecular structure and electronic properties. This study provides a comparative quantum chemical analysis of DX and HC to clarify how these differences influence hydrogen bonding strength, reactivity, and their potential interactions with the glucocorticoid receptor (GR). Optimized geometries, natural bond orbital (NBO) analyses, frontier molecular orbitals (FMO), global reactivity descriptors, and average local ionization energy (ALIE) calculations demonstrate that DX exhibits greater polarity and electrophilic character compared to HC. These differences help explain the stronger receptor binding affinity observed for DX. Indeed, notably, despite the inherent limitations of gas-phase DFT calculations compared to experimental X-ray data, the theoretical results exhibit good agreement with experimental observations, suggesting the reliability of the computational approach in predicting molecular interactions within the GR active site.
Methods
All quantum chemical calculations were performed using density functional theory (DFT) with the B3LYP functional and 6-311++G(d,p) basis set. Structural optimization, FMO analysis, global reactivity descriptors, and dipole moment evaluations were carried out in Gaussian 09. NBO analysis was performed with NBO 5.0. Average local ionization energy (ALIE) surfaces were generated using Multiwfn 3.8, and molecular visualizations were produced with GaussView 5.0.
期刊介绍:
The Journal of Molecular Modeling focuses on "hardcore" modeling, publishing high-quality research and reports. Founded in 1995 as a purely electronic journal, it has adapted its format to include a full-color print edition, and adjusted its aims and scope fit the fast-changing field of molecular modeling, with a particular focus on three-dimensional modeling.
Today, the journal covers all aspects of molecular modeling including life science modeling; materials modeling; new methods; and computational chemistry.
Topics include computer-aided molecular design; rational drug design, de novo ligand design, receptor modeling and docking; cheminformatics, data analysis, visualization and mining; computational medicinal chemistry; homology modeling; simulation of peptides, DNA and other biopolymers; quantitative structure-activity relationships (QSAR) and ADME-modeling; modeling of biological reaction mechanisms; and combined experimental and computational studies in which calculations play a major role.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


