Meire Y. Kawamura, David J. Ponting, Chris G. Barber, Michael J. Burns
{"title":"Computational mechanistic study on N-nitrosation reaction of secondary amines","authors":"Meire Y. Kawamura, David J. Ponting, Chris G. Barber, Michael J. Burns","doi":"10.1007/s00894-025-06520-7","DOIUrl":null,"url":null,"abstract":"<div><h3>Context</h3><p>The presence of potentially carcinogenic nitrosamines in drugs has been a worldwide concern, driving strategies to control or mitigate their formation to protect patient health. Understanding the critical factors for <i>N</i>-nitrosation, such as mechanisms and energy barriers, enhances the risk assessment process to understand potential nitrosamine formation. Evaluation of the structural impact of amines on the <i>N</i>-nitrosation rate in the presence of nitrites and acidic media is of great interest to pharmaceutical companies assessing the risk of nitrosamine drug substance–related impurities. A range of secondary amines was explored using DFT calculations to assess the impact of electronic and steric effects on activation energy. <i>Asym</i>-N<sub>2</sub>O<sub>3</sub> was selected as the nitrosating agent since its reaction was shown to be favorable following screening of pathways employing nitrosyl chloride, nitrous acid, <i>asym</i>-N<sub>2</sub>O<sub>3</sub>, <i>sym</i>-N<sub>2</sub>O<sub>3</sub>, and <i>trans-cis</i>-N<sub>2</sub>O<sub>3</sub>. The relatively low activation energies obtained for all amines indicate the reaction is very likely to occur if the reactive components encounter, even for amines with sterically hindered and electron-withdrawing groups. Understanding the interaction between the amine and nitrosating agent is therefore the defining factor in the risk of formation of more complex nitrosamines within drugs.</p><h3>Methods</h3><p>Calculations were performed using the Gaussian-16 program. The B3LYP-D3/def2-TZVP level of theory was applied for structure optimizations. The IEF-PCM implicit model was used for the solvent effect. Intrinsic reaction coordinate calculations were carried out to connect the transition state with the associated minimum.\n</p><h3>Graphical Abstract</h3>\n<div><figure><div><div><picture><source><img></source></picture></div></div></figure></div></div>","PeriodicalId":651,"journal":{"name":"Journal of Molecular Modeling","volume":"31 11","pages":""},"PeriodicalIF":2.5000,"publicationDate":"2025-10-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Modeling","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s00894-025-06520-7","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Context
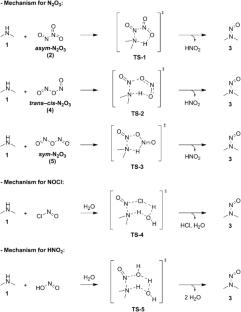
The presence of potentially carcinogenic nitrosamines in drugs has been a worldwide concern, driving strategies to control or mitigate their formation to protect patient health. Understanding the critical factors for N-nitrosation, such as mechanisms and energy barriers, enhances the risk assessment process to understand potential nitrosamine formation. Evaluation of the structural impact of amines on the N-nitrosation rate in the presence of nitrites and acidic media is of great interest to pharmaceutical companies assessing the risk of nitrosamine drug substance–related impurities. A range of secondary amines was explored using DFT calculations to assess the impact of electronic and steric effects on activation energy. Asym-N2O3 was selected as the nitrosating agent since its reaction was shown to be favorable following screening of pathways employing nitrosyl chloride, nitrous acid, asym-N2O3, sym-N2O3, and trans-cis-N2O3. The relatively low activation energies obtained for all amines indicate the reaction is very likely to occur if the reactive components encounter, even for amines with sterically hindered and electron-withdrawing groups. Understanding the interaction between the amine and nitrosating agent is therefore the defining factor in the risk of formation of more complex nitrosamines within drugs.
Methods
Calculations were performed using the Gaussian-16 program. The B3LYP-D3/def2-TZVP level of theory was applied for structure optimizations. The IEF-PCM implicit model was used for the solvent effect. Intrinsic reaction coordinate calculations were carried out to connect the transition state with the associated minimum.
期刊介绍:
The Journal of Molecular Modeling focuses on "hardcore" modeling, publishing high-quality research and reports. Founded in 1995 as a purely electronic journal, it has adapted its format to include a full-color print edition, and adjusted its aims and scope fit the fast-changing field of molecular modeling, with a particular focus on three-dimensional modeling.
Today, the journal covers all aspects of molecular modeling including life science modeling; materials modeling; new methods; and computational chemistry.
Topics include computer-aided molecular design; rational drug design, de novo ligand design, receptor modeling and docking; cheminformatics, data analysis, visualization and mining; computational medicinal chemistry; homology modeling; simulation of peptides, DNA and other biopolymers; quantitative structure-activity relationships (QSAR) and ADME-modeling; modeling of biological reaction mechanisms; and combined experimental and computational studies in which calculations play a major role.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


