{"title":"Prior knowledge on context-driven DNA fragmentation probabilities can improve de novo genome assembly algorithms.","authors":"Patrick Pflughaupt, Aleksandr B Sahakyan","doi":"10.1186/s12859-025-06267-1","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>De novo genome assembly poses challenges when dealing with highly degraded DNA samples or ultrashort sequencing reads. Probabilistic approaches have been offered to enhance the algorithms, though existing methods rely solely on expected k-meric frequencies in the assemblies, neglecting the broader sequence context that strongly influences DNA fragmentation patterns.</p><p><strong>Results: </strong>Here, we present a proof of concept showing that prior knowledge on sequence context-driven DNA breakage propensities, through the dedicated parameterisation of k-mer assigned breakage probabilities, can be utilised to recover DNA assemblies that originate from fragmentation patterns more likely to have happened. Our approach is beneficial even for read lengths below the common ∼ 25 bp threshold of modern de novo genome assembly algorithms, and well below the threshold used for ultrashort fragments used in ancient DNA research.</p><p><strong>Conclusions: </strong>This work could lay the groundwork for future enhanced de novo genome assembly algorithms, with improved ability to effectively assemble and evaluate ultrashort DNA fragments relevant for cell-free, ancient, and forensic DNA research.</p>","PeriodicalId":8958,"journal":{"name":"BMC Bioinformatics","volume":"26 1","pages":"245"},"PeriodicalIF":3.3000,"publicationDate":"2025-10-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12519797/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s12859-025-06267-1","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
Background: De novo genome assembly poses challenges when dealing with highly degraded DNA samples or ultrashort sequencing reads. Probabilistic approaches have been offered to enhance the algorithms, though existing methods rely solely on expected k-meric frequencies in the assemblies, neglecting the broader sequence context that strongly influences DNA fragmentation patterns.
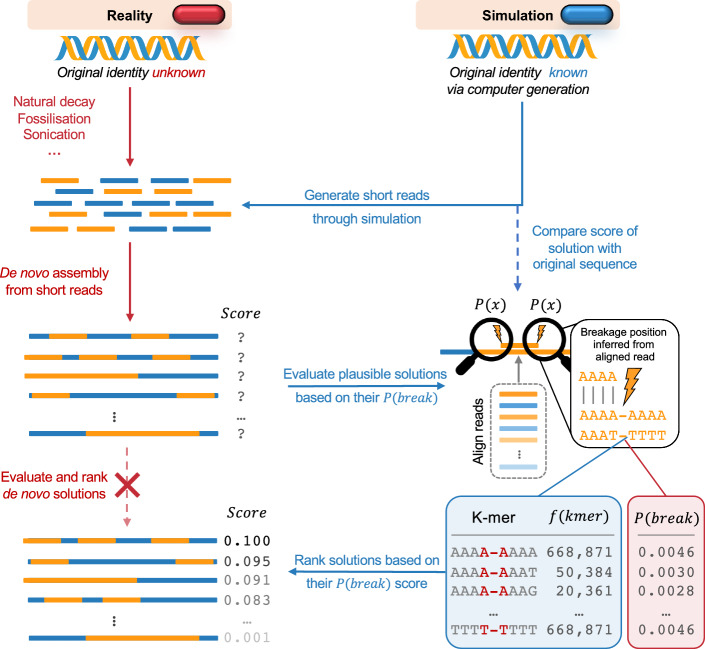
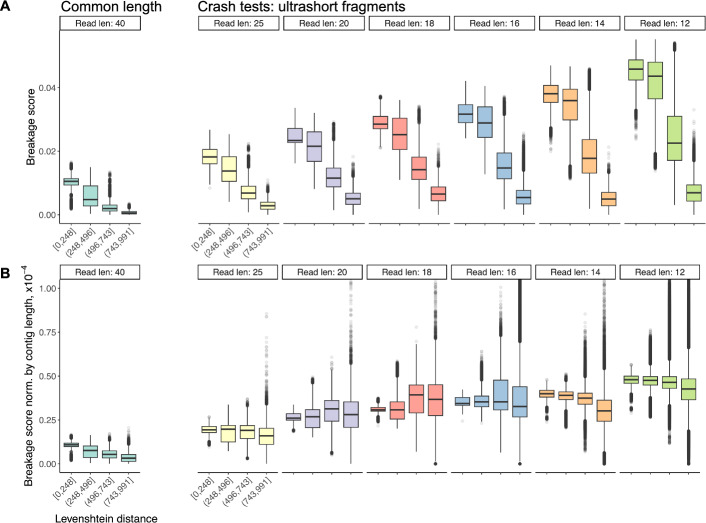
Results: Here, we present a proof of concept showing that prior knowledge on sequence context-driven DNA breakage propensities, through the dedicated parameterisation of k-mer assigned breakage probabilities, can be utilised to recover DNA assemblies that originate from fragmentation patterns more likely to have happened. Our approach is beneficial even for read lengths below the common ∼ 25 bp threshold of modern de novo genome assembly algorithms, and well below the threshold used for ultrashort fragments used in ancient DNA research.
Conclusions: This work could lay the groundwork for future enhanced de novo genome assembly algorithms, with improved ability to effectively assemble and evaluate ultrashort DNA fragments relevant for cell-free, ancient, and forensic DNA research.
期刊介绍:
BMC Bioinformatics is an open access, peer-reviewed journal that considers articles on all aspects of the development, testing and novel application of computational and statistical methods for the modeling and analysis of all kinds of biological data, as well as other areas of computational biology.
BMC Bioinformatics is part of the BMC series which publishes subject-specific journals focused on the needs of individual research communities across all areas of biology and medicine. We offer an efficient, fair and friendly peer review service, and are committed to publishing all sound science, provided that there is some advance in knowledge presented by the work.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


