Investigation of the Effect of Framework Flexibility on CO2 Adsorption in SIFSIX-3-Cu Using a Machine-Learned Force Field
IF 3.2
3区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
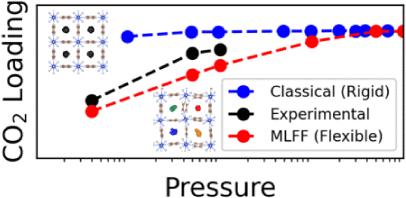
Metal–organic frameworks (MOFs) offer promise as selective CO2 sorbents, but successful MOF sorbent materials need high CO2 binding affinity and selectivity for CO2 over water. This work focuses on the use of machine-learned force fields (MLFFs) to model CO2 adsorption in flexible MOFs, with a focus on SIFSIX-3-Cu, an anion-pillared MOF known for its high CO2 affinity. A preliminary high-throughput screening of over 900 anion-pillared MOFs was performed using rigid UFF+DDEC6 force fields to predict zero-loading heats of adsorption for CO2 and H2O. SIFSIX-3-Cu was selected for further computational study due to its predicted CO2 heat of adsorption and experimental relevance. A DeePMD-based MLFF was trained to reproduce DFT (PBE+D3) energies and forces, with an iterative sampling scheme combining molecular dynamics, geometry optimization, random geometric insertion, and NVT Monte Carlo-based configuration generation to capture both attractive and repulsive regions of the potential energy surface. Flexibility of the MOF was explicitly included, contrasting with previous models that approximated the MOF as rigid. Hybrid Monte Carlo/molecular dynamics (MC/MD) simulations with the MLFF produced CO2 adsorption isotherms in good agreement with experimental data at direct air capture (DAC) pressures (e.g., 40 Pa), in contrast to previous overestimations of CO2 sorption by models with rigid structures. Bond and angle histogram analysis showed that MOF flexibility increased the variance of fluorine–fluorine diagonal distances at adsorption sites, resulting in a lower predicted sorption for flexible, asymmetric SIFSIX-3-Cu pore geometries compared to the rigid, symmetric DFT-optimized SIFSIX-3-Cu pore geometry. A detailed description of flexibility afforded by the MLFF resulted in an accurately predicted CO2 uptake (0.88 mmol/g) at low pressure (40 Pa) compared to the experimentally measured value (1.24 mmol/g). These results underscore the importance of including framework flexibility when modeling adsorption phenomena in MOFs, particularly for low-pressure applications.
利用机器学习力场研究框架柔性对sif6 -3- cu吸附CO2的影响
金属有机骨架(MOF)是一种有前途的选择性CO2吸附剂,但成功的MOF吸附剂材料需要具有高的CO2结合亲和力和对CO2在水中的选择性。这项工作的重点是使用机器学习力场(MLFFs)来模拟柔性MOF中的CO2吸附,重点是sif6 -3- cu,一种以其高CO2亲和力而闻名的阴离子支柱MOF。使用刚性UFF+DDEC6力场对900多种阴离子柱mof进行了初步高通量筛选,以预测CO2和H2O的零负载吸附热。sif6 -3- cu由于其预测的CO2吸附热和实验相关性而被选择进行进一步的计算研究。通过结合分子动力学、几何优化、随机几何插入和NVT蒙特卡罗配置生成的迭代采样方案,训练基于deepmd的MLFF来再现DFT (PBE+D3)的能量和力,以捕获势能表面的吸引和排斥区域。与之前将MOF近似为刚性的模型相比,明确地包括了MOF的灵活性。在直接空气捕获(DAC)压力(例如,40 Pa)下,MLFF的混合蒙特卡罗/分子动力学(MC/MD)模拟得出的二氧化碳吸附等温线与实验数据非常吻合,这与之前刚性结构模型对二氧化碳吸附的高估形成了鲜明对比。键和角直方图分析表明,MOF柔韧性增加了吸附位点上氟-氟对角线距离的方差,导致柔性的、不对称的sif6 -3- cu孔几何比刚性的、对称的dft优化的sif6 -3- cu孔几何的预测吸附量更低。与实验测量值(1.24 mmol/g)相比,MLFF提供的灵活性的详细描述导致在低压(40 Pa)下准确预测CO2吸收率(0.88 mmol/g)。这些结果强调了在模拟mof中的吸附现象时包括框架灵活性的重要性,特别是在低压应用中。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

The Journal of Physical Chemistry C
化学-材料科学:综合
CiteScore
6.50
自引率
8.10%
发文量
2047
审稿时长
1.8 months
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


